
Translate this page into:
Ectopic Anterior Mediastinal Pathology in the Chest: Radiologic-pathologic Correlation of Unexpected Encounters with the “Terrible Ts”
Address for correspondence: Dr. Oleg Epelbaum, Division of Pulmonary, Critical Care, and Sleep Medicine, Westchester Medical Center, New York Medical College, 100 Woods Road, Macy Pavilion, Valhalla, NY 10595, USA. E-mail: oleg.epelbaum@wmchealth.org
-
Received: ,
Accepted: ,
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
The complex embryology of the anterior mediastinum makes it home to an array of primary neoplasms tied to the presence of the thyroid and thymus glands in that compartment. While the occurrence of ectopic thyroid deposits in the extramediastinal thorax has not been convincingly established, the other three “Ts” of the classic “4T” mnemonic for the differential diagnosis of an anterior mediastinal mass have occurred in the lung parenchyma, pleural space, and endobronchially as primary tumors. Finding any of the three lesions – thymoma, teratoma, or B-cell lymphoma – in the chest outside the mediastinum is very unusual, but that possibility exists. Herein, we illustrate examples of this rare phenomenon.
Keywords
Anterior mediastinum
lymphoma
pleural
teratoma
thymoma


INTRODUCTION
Translated from the Greek, mediastinum literally means “that which stands between.” In the human, this term signifies the space between the lungs containing, among other structures, the great vessels, thymus gland, pericardium, and esophagus. The borders of the mediastinum as a whole are the thoracic inlet superiorly, the diaphragm inferiorly, the sternum anteriorly, and the vertebral column posteriorly. Historically, the mediastinum has been subdivided into either three or four compartments depending on the classification system to group mediastinal pathology by location. One such system proposed recently refers to the traditional anterior mediastinum as the prevascular compartment with its posterior border formed by the anterior aspect of the pericardium.[1] Of the three compartments, the anterior (prevascular) one accounts for the greatest breadth of pathology, in part because the thymus gland located there has been implicated in a spectrum of neoplastic processes. The classic mnemonic for the differential diagnosis of an anterior mediastinal mass is the proverbial “4 Ts:” thymoma, teratoma, “terrible” lymphoma, and thyroid. While clinicians are accustomed to encountering these entities in the anterior mediastinum, it may not be widely recognized that, very rarely, they can be found in the thorax away from the mediastinum. Of the “4 Ts,” thyroid tissue in the lung or pleural space has not been conclusively established to occur outside the context of metastatic thyroid malignancy.[2] On the other hand, thymoma, teratoma, and B-cell lymphoma all unquestionably, albeit uncommonly, do arise as primary tumors in the extramediastinal thorax. Herein, we illustrate the occurrence of these three entities each in a different location within the chest: the pleural space, the lung parenchyma, and as an endobronchial lesion.
INTRAPLEURAL THYMOMA
A 45-year-old man originally from Ghana was evaluated as an outpatient for oral candidiasis. His physical examination and routine laboratory studies were unremarkable. Testing for the human immunodeficiency virus was negative. Posteroanterior (PA) chest radiograph (CXR) showed a large ovoid density occupying the right mid- and lower-lung fields and silhouetting the right cardiac border [Figure 1a]. Subsequent computed tomography (CT) of the chest performed following the administration of intravenous contrast confirmed the presence of a large mass located in the territory of the right middle and lower lobes with areas of hypodensity consistent with necrosis [Figure 1b and c]. The adjacent lung parenchyma appeared normal. CT-guided core needle biopsies of the lesion showed dense lymphocytic infiltration with an admixture of larger cells with fine chromatin. Immunohistochemistry (IHC) determined that the lymphocyte population consisted entirely of T-cells based on strong positivity for CD3, CD43, and TdT and an elevated Ki-67 index while the other cell type stained for cytokeratins AE1/AE3 and p63, suggesting an epithelial origin. The need to exclude T-cell lymphoma prompted cytogenetic analysis for clonal T-cell receptor gene rearrangement, which was negative for monoclonality. Definitive diagnosis was pursued by means of resection through open thoracotomy. On entering the right thoracic cavity through the video-assisted thoracoscopic approach, it was recognized that this was not a parenchymal but rather a pleural mass with only loose attachments to the anterior mediastinum, which contained grossly and microscopically normal residual thymic tissue. The mass was excised en bloc after conversion to a minithoracotomy. The surgical specimen was a firm, rubbery mass covered by a shiny capsule and measuring 12.5 cm × 11.5 cm × 6.4 cm. Histology confirmed the presence of a mixture of lymphocytes and epithelioid cells separated into lobules by fibrous bands [Figure 1d and e]. This morphology was deemed consistent with thymoma type B1 based on the World Health Organization (WHO) classification. Following an uneventful postoperative course, the patient has remained free of recurrence. No evidence of a parathymic syndrome was ever discovered.
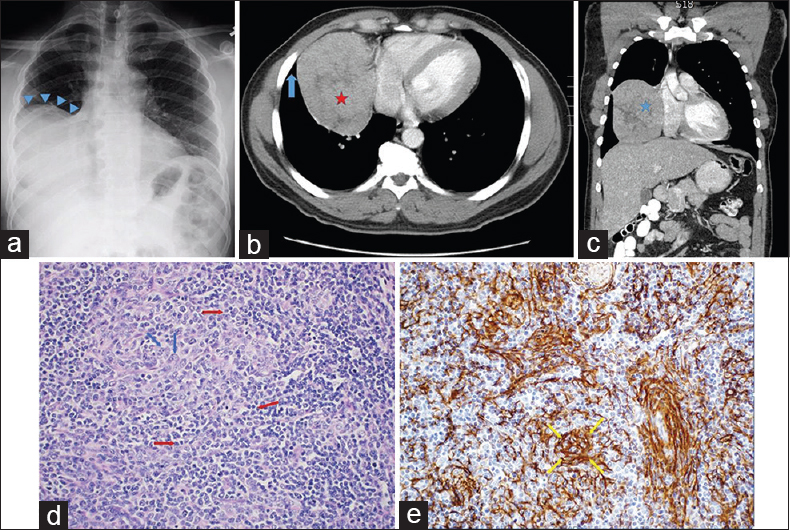
- A 45-year-old otherwise healthy African man undergoing evaluation for oral thrush. (a) Posteroanterior chest radiograph shows a large mass-like density occupying the mid- and lower-right hemithorax (blue arrowheads). (b) Axial contrast-enhanced chest computed tomography image shows a heterogeneous right lung mass (red star) abutting the right atrium. The mass is forming an acute angle with the chest wall (blue arrow), which is more typical of a lung parenchymal rather than intrapleural lesion. (c) Coronal computed tomography reconstruction view depicts the mass in a different projection (blue star). (d) Surgical specimen section stained with H and E shows intermixed lymphocytes (red arrows) and epithelial cells (blue arrows) separated by fibrous septa, morphologically consistent with the diagnosis of thymoma (H and E, ×200). (e) Positive immunohistochemical staining for cytokeratin (yellow arrows) confirms the epithelial origin of the nonlymphoid component (H and E, ×200).
DISCUSSION
The thymus gland is the site of T-lymphocyte maturation and is located in the anterior mediastinal compartment. Thymoma, a potentially malignant neoplasm arising from the epithelial cells of the thymus, is a rare tumor with no gender predilection. According to one estimate, there are roughly 390 cases/year occurring in the United States.[3] The overwhelming majority of thymomas originate at the site of the gland itself and are prone to local invasion, including the pleura, more so than to distant metastasis. A very uncommon scenario (about 4% of cases) is the occurrence of thymoma as a primary tumor in ectopic locations such as the posterior mediastinum, the neck, and the lung parenchyma.[45] The theory behind these cases is the aberrant deposition of thymic tissue during embryogenesis as it descended from the third pharyngeal pouch to the mediastinum along the neck.[6] Most mediastinal thymomas are either discovered incidentally or because of symptoms related to involvement of adjacent structures such as the superior vena cava syndrome. Approximately half of thymomas are associated with paraneoplastic (parathymic) phenomena, most commonly myasthenia gravis, which are likely mediated through autoimmunity and thus highlight the role of the gland in the immune response.[7] The two systems that are used most widely to stage thymomas are Masaoka staging based on degree of invasion from complete containment within the capsule to metastasis and the WHO classification based on histology ranging from bland to carcinoma.[4] Both systems correlate well with prognosis. CT is the standard imaging modality used in the evaluation of anterior mediastinal masses, including thymomas, and can be used to guide core needle biopsy for tissue diagnosis. Patients with nondiagnostic percutaneous biopsies can then undergo operative sampling. Complete surgical resection to the extent possible is the preferred treatment approach with chemotherapy generally reserved for inoperable or recurrent cases.[4]
Contemporary descriptions of isolated pleural thymomas are limited to a series of eight patients by Moran et al.,[8] and five subsequent case reports.[910111213] An issue raised in the 1992 series[8] about prior reports of pleural thymomas is that on review of those antecedent cases, it was determined that all of them could be considered extensions from a mediastinal primary. Four of the five reports[9111213] published since have explicitly stated that, as in our case, there was no concurrent mediastinal disease in continuity with the pleural lesion. Understandably, this diagnosis was not suspected in any of the individual cases even if the mass was identified preoperatively as intrapleural. More common entities such as mesothelioma and solitary fibrous tumor of the pleura were favored initially.[912] The patients’ demographics and clinical presentations have been quite variable though none was diagnosed with a parathymic syndrome. Whenever tumor size was reported, these pleural thymomas measured at least 10 cm in greatest dimension but were amenable to complete excision though encasement of lung tissue and adherence to the hemidiaphragm have been known to complicate resection.[911] Available follow-up information is quite limited, and it remains unclear whether the staging systems used for mediastinal thymoma are applicable to the pleural cases.
INTRAPULMONARY TERATOMA
A 27-year-old woman originally from Ecuador was referred to the pulmonary clinic of our hospital for evaluation of chronic cough productive of yellow sputum. She had been prescribed multiple courses of antibiotics with no improvement. Physical examination and routine laboratory evaluation were both normal. PA and lateral X-ray views of the chest showed a large right upper lobe (RUL) mass containing multiple internal lucencies [Figure 2a and b]. CT of the chest without intravenous contrast demonstrated a mixed-attenuation lesion located anteromedially within the RUL with both solid and cystic areas, as well as punctate calcification and an air crescent [Figure 2c and d]. These findings were deemed compatible with an aspergilloma. The subsequent fungal culture of the sputum yielded Aspergillus versicolor. She was referred to thoracic surgery for resection of a presumed aspergilloma. On entering the right thoracic cavity through open thoracotomy, a firm RUL mass was encountered. There were significant adhesions of the RUL parenchyma to the mediastinum and pulmonary vessels. Right upper lobectomy was performed. Examination of the gross specimen revealed a partially necrotic mass 6 cm in greatest dimension containing structures resembling hair intermixed with a calcific material akin to “chalk.” Microscopic sections demonstrated the presence of skin and sebaceous glands along with cartilage and bronchial epithelium features consistent with a mature cystic teratoma [Figure 2e and f]. Although there was an area of abscess formation within the mass, no hyphal structures were identified, and special stains were negative for fungal forms. When the CT images were reviewed in retrospect, the central area of low attenuation within the mass [white arrow in Figure 2c] measured − 38 to − 94 Hounsfield units, a range consistent with fat density. The patient recovered uneventfully.
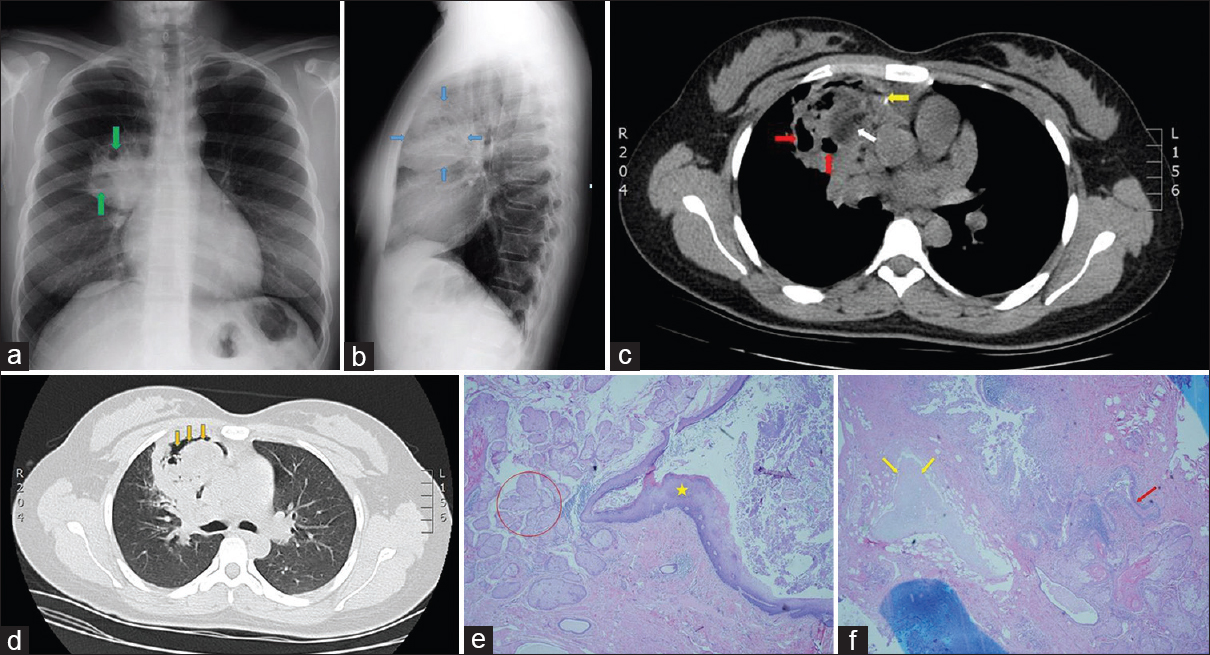
- A 27-year-old woman from Ecuador presenting with chronic productive cough. (a) Posteroanterior chest radiograph shows a large right upper lobe mass with internal lucencies (green arrows). (b) Lateral projection of the chest radiograph localizes the mass to the anterior segment of the right upper lobe (blue arrows). (c) Axial chest computed tomography image without intravenous contrast shows a heterogeneous mass in the anterior right upper lobe containing cystic lucencies (red arrows) and a punctate calcification (yellow arrow). Likewise present is an area of low attenuation measuring fat density (white arrow). (d) Lung window chest computed tomography image depicts an air crescent within the mass (orange arrows). (e) Section from surgical specimen contains ectodermal elements: epidermis (asterisk), and sebaceous glands (red circle); (H and E, ×200). (f) Different area of the mass contains cartilage (yellow arrows) and bronchial epithelium (red arrow), which are derived from mesoderm and endoderm, respectively (H and E, ×200).
DISCUSSION
Mature teratomas are the most common type of germ cell tumor, defined by the presence of well-differentiated tissues derived from at least two of the three embryonic germinal layers: endoderm (e.g., pancreas), mesoderm (e.g., cartilage), and ectoderm (e.g., hair). Dermoid cysts are a subtype of mature teratomas that, as the name suggests, contain cystic structures lined by epidermis and are predominantly composed of ectodermal elements. Several theories have been advanced to explain the occurrence of extragonadal teratomas in general. The favored hypothesis is that they result from aberrant deposition of primitive germ cells during their migration from the yolk sac to the genital ridges along the hindgut, which would explain their typical midline location (i.e., brain, mediastinum).[14] The main competing view is that they arise from pluripotent stem cells. Mature teratomas are usually entirely benign although they can harbor islets of malignant degeneration that should be carefully sought in the pathology specimen, in which case they are termed “teratocarcinomas.” The mediastinum is the most common location of extragonadal germ cell tumors in adults, whereas fewer than 100 cases of lung parenchymal teratomas have been reported since the original description in the 19th century. That number is likely to be somewhat exaggerated by inclusion of patients, in which extension of a mediastinal teratoma is mistaken for a primary intrapulmonary origin. It has been proposed that, developmentally, true intrapulmonary teratomas arise from a primordial focus that initially descended into the mediastinum from the third pharyngeal pouch as part of the thymus but then was aberrantly incorporated into one of the embryos’ lung buds growing from the ventral wall of the foregut.[15] The well-described association of thymic tissue with both mediastinal and intrapulmonary teratomas lends support to the notion that the genesis of mediastinal and by extension, intrapulmonary teratomas is from the embryonic thymus.[16]
Only a minority of patients with an intrapulmonary teratoma will be symptomatic. Among those with symptoms, chest pain, hemoptysis, and cough are the most common complaints.[17] Occasionally, these lesions will contain a coexistent abscess, in which case infectious symptoms may also be present. Hair expectoration (trichoptysis), reported in about 10% of cases, is considered virtually diagnostic for an intrapulmonary location of a teratoma because it implies direct communication with a bronchus.[18] Bronchiectasis has also been described as a complication of endobronchial involvement either as a result of mechanical distortion or as a postobstructive phenomenon.[19]
The majority of intrapulmonary teratomas have been located in the upper lobes, which likely relates to the embryological mechanism underlying their formation. Plain radiography demonstrates a soft-tissue mass often associated with the ipsilateral hilum that may contain calcification although identifiable dental or skeletal structures per se are a rare finding in extragonadal teratomas. An air crescent (Monod sign) such as the one seen in our case is an unusual feature that, when present, has led to preoperative misdiagnosis of the lesion as a mycetoma.[1920] On CT, an intrapulmonary teratoma appears as a well-circumscribed, heterogeneous mass with intermixed solid and cystic components. If areas of soft tissue, fluid, and fat attenuation are seen concurrently within the solid material, there are sufficient grounds for the radiological diagnosis of teratoma, especially when calcification is also present. The presence of fat in our patient's lesion on CT was originally missed. In some cases, an actual fat-fluid level can be discerned, thought to represent the interface between lipid-rich sebum produced by the ectodermal layer and the serous fluid secreted by endodermal elements.[21] This sign is pathognomonic for teratoma but occurs only about 10% of the time.[22]
Surgical resection is recommended for all teratomas in part because of the potential for malignant transformation. Furthermore, in addition to simple mass effect, lesions that contain pancreatic tissue are capable of secreting digestive enzymes that could lead to erosion and rupture into adjacent structures. Postresection recurrence of a mature teratoma has not been described.[18]
ENDOBRONCHIAL LYMPHOMA
A 50-year-old man originally from Malaysia presented to the hospital with subjective fever, cough with occasional streaks of blood, and right-sided chest pain of 4-day duration. Before the onset of these symptoms, he reported feeling well. He was a former smoker, and he had no personal history of tuberculosis or known prior exposure to tuberculosis. He had a history of alcohol abuse. His vital signs were remarkable for a temperature of 101.1°F. Physical examination revealed stigmata of chronic liver disease; cardiopulmonary examination was normal, and there was no palpable lymphadenopathy. Routine laboratory evaluation was significant for a hemoglobin of 10.4 g/dL (reference range: 13.5–17.5) with a mean corpuscular volume of 105 fL (reference range: 80–100) and a platelet count of 60 K/mcL (reference range: 130–400). Serum lactate dehydrogenase level was normal. Bacterial blood cultures returned negative. PA and lateral X-ray views of the chest showed an enlarged right hilum with a peripheral lung consolidation concerning for a postobstructive pneumonia [Figure 3a and b]. Subsequent contrast-enhanced CT of the chest confirmed the presence of a right hilar mass enveloping the right middle lobe (RML) bronchus [Figure 3c] and abutting the right pulmonary artery with encasement of its RUL branch [Figure 3d]. Mediastinal lymphadenopathy (not shown) and a dense RUL posterior segment consolidation with air bronchograms [Figure 3e] were likewise present. On bronchoscopic inspection, the RML bronchus appeared extrinsically narrowed, and an endobronchial mass was seen protruding into the lumen in the right lower lobe at the level of the superior segment bronchus. Needle aspiration of the endobronchial lesion was performed for both cytology and histology samples in addition to endobronchial forceps biopsies. The RUL consolidation was not biopsied on the assumption that it represented postobstructive pneumonia in a febrile patient. The needle specimens were nondiagnostic while forceps biopsies showed bronchial wall infiltrated by round blue cells with marked crush artifact distorting the architecture. IHC revealed positivity for B-cell markers CD20 and CD45, findings suggestive, but not diagnostic, of B-cell lymphoma. The patient then underwent repeat bronchoscopy with additional biopsies of the same endobronchial lesion. Extensive procedural hemorrhage from the endobronchial biopsies precluded sampling of the RUL consolidation. Histology from that procedure confirmed the presence of lymphoma, classified as diffuse large B-cell lymphoma (DLBCL) considering IHC positivity for CD20, CD45, PAX-5, and BCL-2 [Figure 3f and g]. Eighty percent of the cells were positive for Ki-67 nuclear labeling. Staining for T-cell markers was negative. Subsequent bone marrow biopsy showed no evidence of lymphoma. No lymphadenopathy was seen on CT of the abdomen and pelvis. The patient underwent five cycles of R-CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone + rituximab) chemotherapy and is currently in remission.
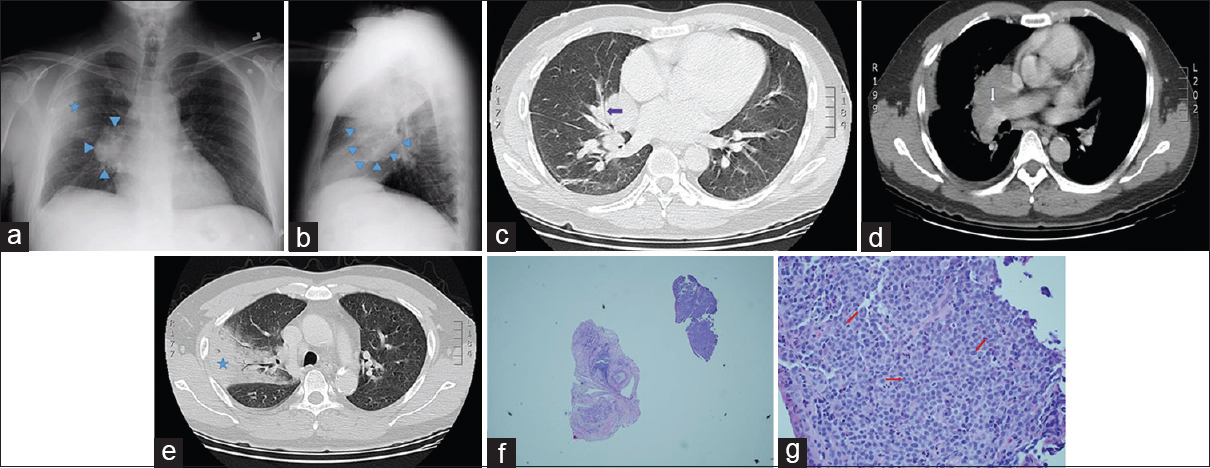
- A 50-year-old Malaysian man presenting with fever, chest pain, and hemoptysis. (a) Posteroanterior plain chest radiograph shows a right hilar mass (blue arrowheads) with an ipsilateral alveolar opacity in the lung periphery (blue star). (b) Lateral plain chest radiograph demonstrates the right hilar mass in a different projection (blue arrowheads). (c) Axial chest computed tomography image with lung windows shows a right hilar mass narrowing the right middle lobe medial segment bronchus (purple arrow). (d) Contrast-enhanced chest computed tomography image demonstrates the right hilar mass encasing the right upper lobe branch of the right pulmonary artery (white arrow). (e) Chest computed tomography image with lung windows shows a right upper lobe posterior segment consolidation clinically presumed to represent postobstructive pneumonia (blue star). (f) Very low-power view of the endobronchial biopsy specimen shows a fragment of tissue (upper right) with a cellular infiltrate and a fragment of benign airway cartilage (center) for comparison. (g) Higher magnification of the diagnostic sample reveals monotonous lymphoid-like cells (red arrows) lacking cohesion and exhibiting a high nuclear: cytoplasmic ratio and prominent nucleoli, a pattern consistent with lymphoma. Immunohistochemical staining established the B-lymphocyte as the neoplastic cell in this tumor (H and E, ×400).
DISCUSSION
The most common type of anterior mediastinal non-Hodgkin's lymphoma (NHL) is primary mediastinal large B-cell lymphoma, a subtype of DLBCL, which accounts for about 4% of all NHL.[23] It is believed to originate from thymic B-lymphocytes. As rare as this entity is, primary pulmonary lymphoma (PPL) is even rarer, representing < 1% of all NHL.[24] In 1963, Saltzstein proposed the following working definition of a “primary lymphocytic tumor of the lung” that remains clinically relevant today: “lymphoma… that originally involves only the lung, or the lung and its regional lymph nodes, and in which there is no evidence of dissemination of the tumor for at least 3 months after the diagnosis is established.”[25]
PPLs arise from so-called bronchus-associated lymphoid tissue (BALT), an aggregation of lymphocytes located in the airway submucosa and induced by antigenic or immunologic stimulation. By far, the most common histology of BALT-type NHL – as much as 90% – is low-grade B-cell lymphoma classified as a type of mucosa-associated lymphoid tissue (MALT) NHL.[26] In fact, the indolent behavior and bland histology of this neoplasm had previously earned it the name “pseudolymphoma” until modern laboratory techniques confirmed its clonal nature. Cases of MALT NHL are often incidental radiological discoveries because the majority of patients are asymptomatic. Patients with the much less common and more aggressive DLBCL variant present with fever, dyspnea, cough, and constitutional symptoms.[24] DLBCL of the lung often occurs in the setting of immunosuppression or immunological disorders.
On plain CXR, PPL manifests as unilateral or bilateral quasi-nodular opacities or mass-like consolidations. CT typically demonstrates a peribronchovascular distribution and the presence of air bronchograms.[27] There may also be an associated ground-glass component as well as thoracic lymphadenopathy. The presence of cavitation or necrosis favors DLBCL over MALT NHL.[28] Competing diagnostic possibilities for this CT pattern include bacterial and nonbacterial infection, alveolar sarcoidosis, granulomatosis with polyangiitis, metastatic or primary lung adenocarcinoma, and organizing pneumonia. Clinical correlation can help narrow the differential diagnosis of these otherwise nonspecific findings.
Definitive diagnosis of PPL is usually established by histological and immunohistochemical examination of a biopsy or resection specimen although the role of bronchoalveolar lavage (BAL) may be underappreciated. It has been shown that an analysis of BAL fluid by polymerase chain reaction for clonal immunoglobulin gene rearrangement has positive and negative predictive values of 82% and 95%, respectively, in cases of PPL.[29]
There is no consensus about the optimal approach to the treatment of BALT NHL. The outcome of these patients is comparatively favorable given 5-year survival rates in excess of 80%, which allows consideration of an initial strategy of watchful waiting in this indolent neoplasm.[26] Historically, many cases of localized unilateral disease have been surgically resected on suspicion of primary lung carcinoma, and this can, in fact, be curative. Radiation with or without adjuvant chemotherapy is an effective medical treatment option for limited disease, whereas chemotherapy including the CD20 monoclonal antibody rituximab is appropriate for advanced disease.[30] Prognosis of DLBCL of the lung is significantly worse according to most series and thus prompt initiation of chemotherapy used for advanced DLCBL is indicated, the standard regimen being R-CHOP.[31323334]
Endobronchial involvement is a particularly rare phenomenon in primary pulmonary NHL, the largest published series of such cases totaling 8 patients.[35] Two distinct forms of the endobronchial spread of PPL have been proposed.[36] Type I pattern, the more common scenario, is characterized by diffuse submucosal infiltration that may be detectable bronchoscopically as airway nodularity but does not result in clinical or radiological evidence of an endobronchial lesion. It has been hypothesized that the cause of Type I involvement could be either centripetal lymphangitic spread from the lung periphery or inward growth of malignantly transformed BALT. Type II pattern refers to the presence of a discrete obstructing mass generally resulting from contiguous invasion by adjacent neoplastic lymph nodes but also originating de novo in exceptional cases. Such focal luminal obstruction usually occurs at the main stem or lobar level and may manifest clinically as cough, hemoptysis, and unilateral wheeze accompanied by evidence of atelectasis on chest imaging. The impact, if any, of endobronchial involvement on treatment or survival in PPL remains to be elucidated.
SUMMARY
Because of the rarity of encountering neoplasms normally located in the anterior mediastinum elsewhere in the chest, they are unlikely to be entertained as initial diagnostic possibilities. Only in the case of a teratoma could a pathognomonic radiological finding lead to consideration of this diagnosis before tissue sampling. The take-home message is that at least 3 of the 4 proverbial “Ts” do occur in other parts of the thorax as primary tumors, so the aim of the foregoing was to increase awareness among radiologists of the existence of such ectopic malignancies.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgments
The authors would like to acknowledge Dr. Zvi Lefkovits, Chairman of the Department of Radiology, Westchester Medical Center, for his help by reviewing the manuscript and offering very valuable advice.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2016/6/1/49/197025
REFERENCES
- A modern definition of mediastinal compartments. J Thorac Oncol. 2014;9(9 Suppl 2):S97-101.
- [Google Scholar]
- Multiple thyroid nodules in the lung: Metastasis or ectopia? Diagn Pathol. 2015;10:61.
- [Google Scholar]
- Epidemiology of thymoma and associated malignancies. J Thorac Oncol. 2010;5(10 Suppl 4):S260-5.
- [Google Scholar]
- Primary intrapulmonary thymoma. A clinicopathologic and immunohistochemical study of eight cases. Am J Surg Pathol. 1995;19:304-12.
- [Google Scholar]
- Schoenwolf GC, ed. Development of the pharyngeal apparatus and face. Larsen's Human Embryology (5th ed). Philadelphia, PA: Churchill Livingstone; 2015. p. :429-72.
- Thymomas presenting as pleural tumors. Report of eight cases. Am J Surg Pathol. 1992;16:138-44.
- [Google Scholar]
- Ectopic pleural thymoma presenting as a giant mass in the thoracic cavity. Ann Thorac Surg. 2007;83:315-7.
- [Google Scholar]
- Ectopic thymoma presenting as a giant intrathoracic tumor: A case report. World J Surg Oncol. 2011;9:66.
- [Google Scholar]
- Ectopic pleural thymoma mimicking a giant solitary fibrous tumour of the pleura. Interact Cardiovasc Thorac Surg. 2012;15:930-2.
- [Google Scholar]
- Ectopic intrapleural thymoma: A rare location in the thoracic cavity. J Surg Case Rep 2016 2016:pii: Rjv166.
- [Google Scholar]
- Why human extragonadal germ cell tumours occur in the midline of the body: Old concepts, new perspectives. Int J Androl. 2007;30:256-63.
- [Google Scholar]
- Intrapulmonary teratoma: A case report and review of the literature. J Thorac Imaging. 1992;7:70-7.
- [Google Scholar]
- A 31-year-old woman with hemoptysis and an intrathoracic mass. Chest. 2010;138:213-9.
- [Google Scholar]
- Endobronchial teratoma associated with bronchiectasis and bronchiolectasis. Thorax. 1968;23:69-76.
- [Google Scholar]
- Mediastinal mature teratoma: Imaging features. AJR Am J Roentgenol. 1997;169:985-90.
- [Google Scholar]
- Mediastinal germ cell tumors: A radiologic-pathologic review. Eur Radiol. 2001;11:1925-32.
- [Google Scholar]
- Pulmonary malignant lymphomas and pseudolymphomas: Classification, therapy, and prognosis. Cancer. 1963;16:928-55.
- [Google Scholar]
- Primary pulmonary lymphoid lesions: Radiologic and pathologic findings. Radiographics. 2016;36:53-70.
- [Google Scholar]
- Clonality analysis of alveolar B lymphocytes contributes to the diagnostic strategy in clinical suspicion of pulmonary lymphoma. Blood. 2004;103:3208-15.
- [Google Scholar]
- Addition of rituximab to chlorambucil produces superior event-free survival in the treatment of patients with extranodal marginal-zone B-cell lymphoma: 5-year analysis of the IELSG-19 randomized study. J Clin Oncol. 2013;31:565-72.
- [Google Scholar]
- Primary pulmonary lymphomas. A clinicopathologic analysis of 36 cases. Cancer. 1984;54:1397-406.
- [Google Scholar]
- Primary pulmonary lymphomas. A clinical study of 70 cases in nonimmunocompromised patients. Chest. 1993;103:201-8.
- [Google Scholar]
- Non-Hodgkin's lymphoma presenting as an endobronchial tumor: Report of eight cases and literature review. Am J Hematol. 2008;83:416-9.
- [Google Scholar]
- Endobronchial involvement with non-Hodgkin's lymphoma. A clinical-radiologic analysis. Cancer. 1986;57:1750-5.
- [Google Scholar]




