
Translate this page into:
Cross-sectional Imaging Features of Primary Retroperitoneal Tumors and Their Subsequent Treatment
-
Received: ,
Accepted: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Basically malignant tumors in the retroperitoneal region arise from a heterogeneous group of tissues: mesodermal, neurogenic, germ cell, and lymphoid. Although rare, benign tumors and cystic masses can be also encountered in retroperitoneal space. Developments in computed tomography (CT) and magnetic resonance imaging (MRI) have contributed to both diagnosis and staging of the retroperitoneal tumors. High spatial resolution and superiority in calcification make CT indispensable; on the other hand, MRI has a better soft-tissue contrast resolution which is essential for the assessment of vascular invasion and tissue characterization. The aim of this article is to review the CT and MRI features of retroperitoneal tumors and their subsequent management.
Keywords
Computed tomography
diagnosis
magnetic resonance imaging
retroperitoneal neoplasms
treatment

INTRODUCTION

The retroperitoneal space, located between posterior parietal peritoneum and transversalis fascia, begins from the diaphragm cranially and ends in the pelvis caudally. This space is basically divided into four regions: The perirenal, anterior pararenal, posterior pararenal, and the great vessel spaces.[1]
Primary retroperitoneal tumors (PRTs), arising in this space, are a heterogeneous group of tumors that can be encountered either in benign or in malignant form. Seventy-eighty percent of all PRTs are malignant in nature and these tumors cover 0.1–0.2% of all malignancies.[2]
Most PRTs have a mesodermal origin, and liposarcoma (LS), leiomyosarcoma (LMS), and undifferentiated pleomorphic sarcoma together constitute greater than 80% of primary retroperitoneal sarcomas. The remaining PRTs originate predominantly from the nervous system.[3] PRTs are usually detected as large masses since the loose connective tissue in the retroperitoneum has an inadequate barrier function against tumoral infiltration.
Computed tomography (CT) and magnetic resonance imaging (MRI) are not only helpful in characterizing the mass lesions in the retroperitoneal space but also effective in determining the extent of the disease. CT is excellent for assessing calcification; on the other hand, MRI has a better soft-tissue contrast, which facilitates staging. Another advantage of MRI is its potential role of determining vascular invasion. Despite developments in multidetector CT technology and new MRI sequences, it is sometimes impossible to make a definitive diagnosis just based on cross-sectional imaging, as some radiological features of PRTs are non-specific and they overlap with each other. Histopathologic evaluation undoubtedly is the inevitable procedure in diagnosis and differential diagnosis in a PRT.[4]
Complete surgical resection is the only potential curative management option for retroperitoneal sarcomas; however, local recurrence occurs in a large proportion of patients and it is responsible for as many as 75% of sarcoma-related deaths.[5]
In this article, we aim to present the CT and MRI features of many PRTs which were confirmed histopathologically under the headings of benign, malignant mesodermal, neurogenic, germ cell, and lymphoid tumors and to review the treatment methods.
BENIGN TUMORS
Lymphangioma
Lymphangioma constitutes 1% of all PRTs and is more commonly detected in men.[4] Although most patients are asymptomatic, it can present with pain and abdominal distention. Lymphangioma results from failure of fusion of retroperitoneal lymphatic tissue with the main lymphatic system. Thin-walled unilocular or multilocular cystic masses are characteristic CT findings in lymphangioma. Density varies from water to fat due to the lipid content of chylous lymphatic fluid [Figure 1].[6] On MRI, a typical lymphangioma has low signal intensity on T1-weighted (T1W) images and high signal intensity on T2-weighted (T2W) images; however, signal intensity may change depending on the amount of lipid present [Figure 2]. Because of the aqueous content, it is relatively easy to distinguish lymphangioma from other solid PRTs. Septa within the lesion, lack of fluid in dependent recesses, and compression of intestinal loops help in differentiating lymphangioma from ascites.
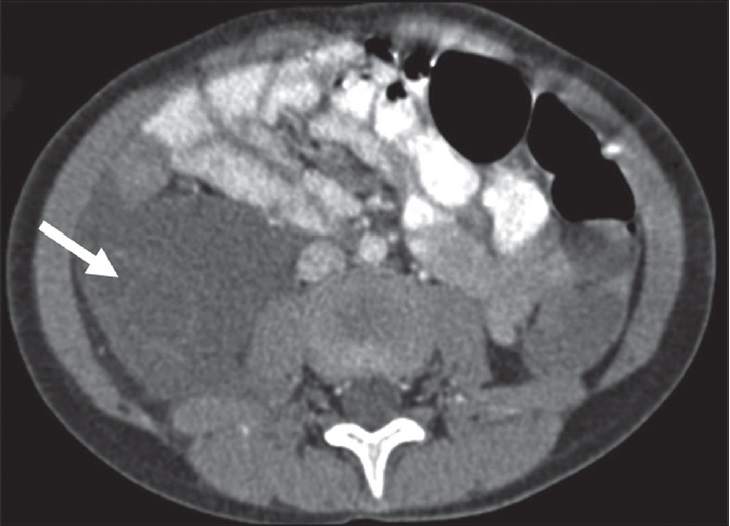
- 5-year-old girl with right-side abdominal distension diagnosed as cystic lymphangioma on surgery. Post-contrast axial CT image shows a smooth-contoured retroperitoneal mass lesion with fluid density. Note that there are also thin enhancing septa (arrow) in the caudal portion of lymphangioma.
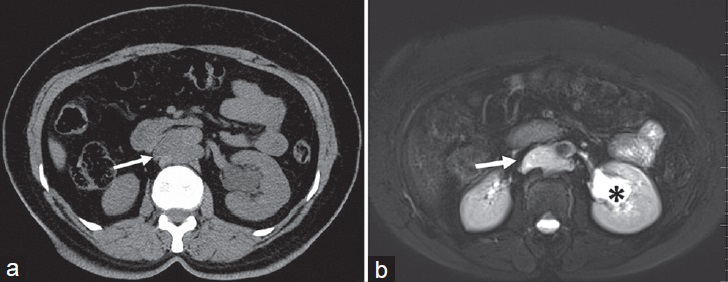
- 25-year-old woman diagnosed with lymphangioma. (a) Axial CT, (b) axial T2W fat-suppressed images show a well-defined cystic mass in the retroaortic region (arrows). There is also parapelvic cyst in the left kidney (asterisk). Note that due to chylous content, T2 signal intensity is slightly decreased with respect to the left kidney cyst (b).
The first treatment of choice in lymphangioma is complete surgical excision. Image-guided percutaneous catheter drainage coupled with sclerotherapy is another alternative treatment method. The long-term prognosis is excellent and no recurrence is expected if complete excision has been achieved.[7]
Lipoma
Lipoma is a benign mesenchymal tumor with fat content resembling that of normal adipose tissue. Lipoma in the retroperitoneum is rare and represents about 2.9% of all PRTs.[8] The appearance of lipoma may be similar to that of an LS; however, LS has thicker, irregular, and nodular septa that demonstrate enhancement in post-contrast images. Also, lipoma is less common than LS in the retroperitoneum.[91011]
Lipoma has been characterized as a homogeneous fatty mass on CT and with a signal intensity identical to that of normal fat in all pulse sequences on MRI [Figure 3].[12] Lipoma in the retroperitoneal compartment shows homogeneous signal reduction in all fat-suppressed MRI sequences and the mass lesion does not enhance in post-contrast images.
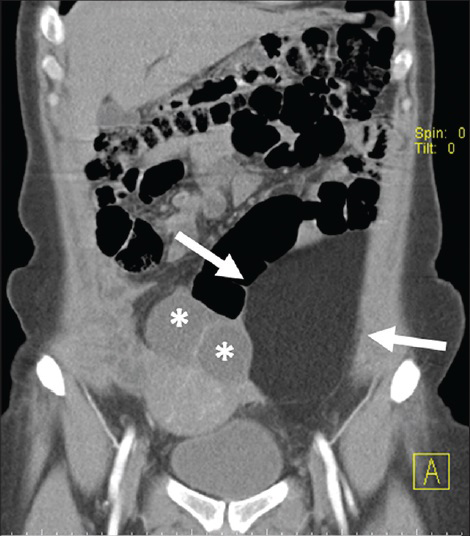
- 43-year-old woman diagnosed with lipoma. Post-contrast CT image shows a well-circumscribed solid mass lesion with homogeneous fat attenuation, in contact with the left external iliac artery. The solid lesion causes displacement of left ovary and uterus due to the mass effect. Note that no thick, irregular, or nodular septa are seen in contrast-enhanced images (arrows). There are also medially displaced cysts originating from the left ovary (asterisks).
Treatment for retroperitoneal lipomas is surgical removal. In most cases, surgical resection is easily performed since the capsule that surrounds the tumor presents a clear cleavage plane with neighboring structures. Radical resection of the lesion should be performed if possible, as co-occurrence of LS is not an uncommon finding, which may lead to the possibility of locoregional relapse.
Angiomyolipoma
Although kidneys are the common predilection site for angiomyolipoma (AML), retroperitoneum, solid organs, skin, and gynecologic tract can be affected in rare cases.[11] This tumor contains predominantly adipocytes, varying amounts of vessels, and smooth muscle cells. Either detected as a sporadic condition or in association with tuberous sclerosis, AMLs are predominantly seen in younger females. Patients may be asymptomatic or may present with retroperitoneal hemorrhage. The bleeding tendency depends on the angiogenic ingredient of the tumor that includes irregular and aneurysmatic blood vessels.[131415] The risk factors that cause hemorrhage are the size of the tumor, the grade of angiogenic component, and the accompanying tuberous sclerosis.[16] Main indications for treatment of patients with renal AML are the tumor size (greater or equal to 4 cm) and presence of symptoms.[15] Treatment options include surgery and arterial embolization. The advantages of arterial embolization over surgery are avoidance of bleeding complications and preservation of the renal parenchyma.[17]
On CT and MRI, small AMLs are homogeneous; however, larger tumors are heterogeneous soft-tissue masses that characteristically contain a large volume of hyperenhancing vascular soft tissue [Figure 4].[3] LSs may resemble AMLs due to presence of large amount of fat; however, hemorrhage, enlarged vessels, and aneurysms are helpful in distinguishing AML from LS.[11] The presence of a tumoral vessel extending into the renal cortex at the site of tumor contact strongly favors the diagnosis of AML, whereas calcifications strongly suggest LS.[18]
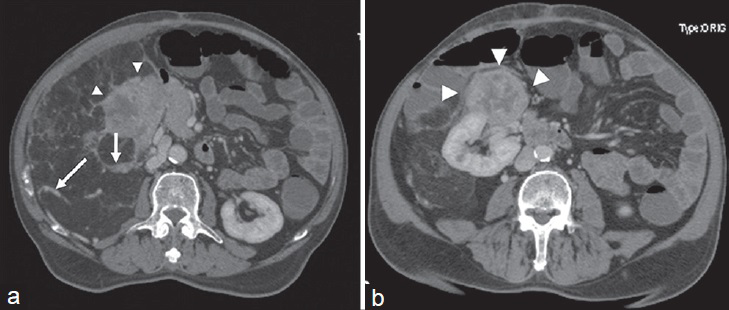
- 54-year-old man diagnosed with angiomyolipoma. (a and b) Post-contrast axial CT images demonstate a gross right pararenal mass lesion containing adipose tissue compatible with angiomyolipoma. Heterogeneous enhancing septa and high attenuation areas are seen (arrows). Note that there is also a hypervascular soft-tissue component originating from the anterior portion of the right kidney, which helps in ruling out a perirenal liposarcoma (arrowheads).
MALIGNANT MESODERMAL TUMORS
Liposarcoma
Liposarcoma (LS), the most common mesodermal malignant tumor, covers 33% of all primary retroperitoneal sarcomas.[2] LSs are usually seen in the 5th and 6th decades of life. Pathologically, LSs are divided into four catagories based on increasing order of malignant potential as follows: Well-differentiated, myxoid, pleomorphic, and round cell subtypes.[919]
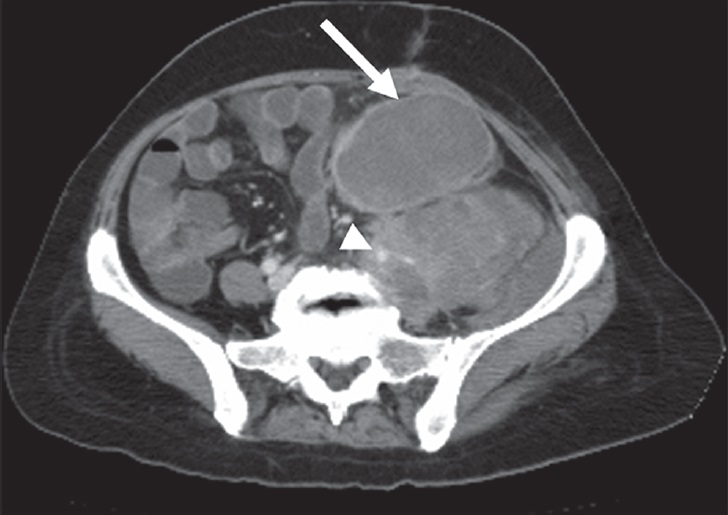
Well-differentiated LS is a predominantly hypoattenuating solid mass on CT images because of its high fat content [Figure 5]. On MR imaging, well-differentiated LS shows intermediate signal intensity on T2W and high signal intensity on T1W images. There is also loss of fat signal on fat-suppressed MRI sequences. Rarely, well-differentiated LS can transform into an aggresive form called dedifferentiated LS. Dedifferentiated LSs are heterogeneous tumors containing both fat and solid components and show a lack of clear delineation between solid and fat components.[3] Calcification in solid component is a sign of dedifferentiation [Figure 6].[11]
- 44-year-old man with well-differentiated liposarcoma. Axial post-contrast CT image demonstrates that the lesion contains heterogeneous fatty content. There are thick septa (arrowheads) that show soft-tissue attenuation.

- 59-year-old woman with recurrent dedifferentiated liposarcoma in the left retroperitoneal region. In her first operation, an 8.8 × 5.5 × 3.5 cm tumor with spleen and left kidney was resected due to visceral invasion. One year later, in the follow-up CT scan, a heterogeneously enhancing solid mass with calcification was seen in the operated area (arrows), which is compatible with locoregional recurrence.
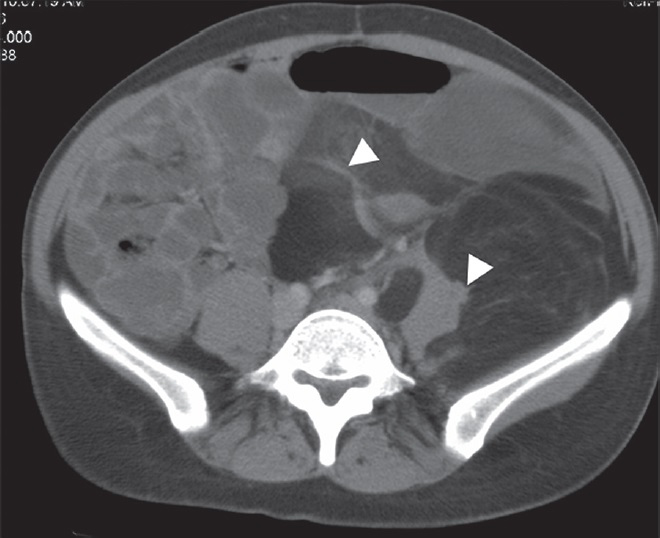
Myxoid LS is seen in a younger population compared to well-differentiated LS. On CT, the mass is more heterogeneous and has a higher attenuation than that of well-differentiated LS. Due to the myxoid stroma, myxoid LSs are seen as cystic mass lesions on CT and MRI [Figure 7]. Heterogeneous contrast enhancement is seen in post-contrast series.[34]
- 63-year-old man diagnosed with myxoid liposarcoma. CT scan shows the mass lesion pushing the left kidney laterally. Body and tail of the pancreas are also infiltrated. Note that myxoid liposarcoma is seen in cystic form due to myxoid stroma. Only a little fat is seen within the lesion (arrow).
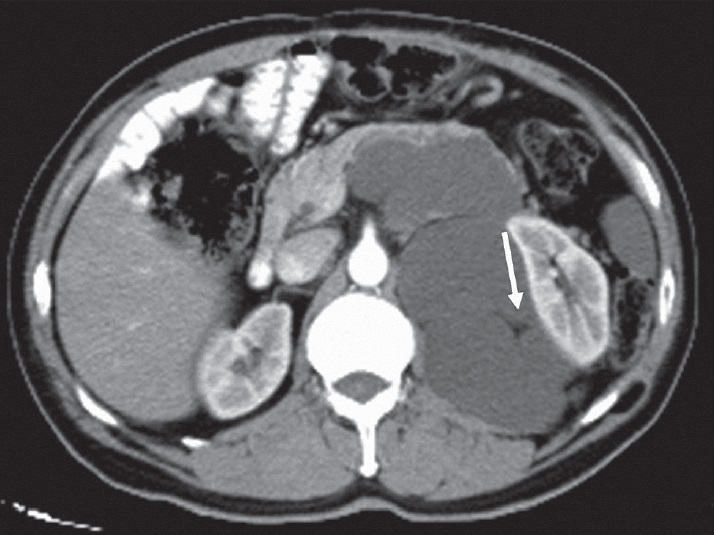
Pleomorphic LS, which is the least common LS variant, is seen as a heterogeneous enhancing mass with necrotic areas and due to small amount of fat, it is not easily distinguished from other solid tumors [Figure 8].
- 62-year-old woman with pleomorphic liposarcoma in the left perirenal area. Axial post-contrast CT image depicts a solid soft-tissue mass lacking macroscopic fat. Note that contrast enhancement increases compared to well-differentiated liposarcoma (arrow).
Although it is not always possible to distinguish each subtype of LS radiologically, there are some tips that may allow radiologist to predict the possible histological subtype. First, retroperitoneal LS is a disease of elders, but myxoid LS is seen in relatively younger ages. Second, an LS in the retroperitoneum is most likely to be well differentiated because well-differentiated LS is the most common variant whereas pleomorphic LS is the least common subtype in the retroperitoneum. Third, well-differentiated LSs contain mature adipose tissue with imaging features that may be indistinguishable from those of normal fat or lipoma. However, pleomorphic LS is commonly seen as a nonspecific soft-tissue mass with little or no visible fat. On the other hand, myxoid LSs contain a high percentage of water and necrosis, which causes a cystic appearance on MR images. Although myxoid LSs are seen as cystic lesions, enhancement after contrast media administration proves that these lesions are “pseudocysts” rather than a true cyst, which was earlier described by Song et al.[19] Lastly, calcification within the lesion is highly suggestive of dedifferentiated LS.[31119]
Surgery is the major treatment option for both primary and recurrent retroperitoneal LSs. Although the goal of the surgical intervention should be complete resection without leaving any residue in the retroperitoneal compartment, large tumor sizes as well as the involvement of critical structures create the main obstacle for complete surgical removal. Therefore, the practical aim of surgery in LS is wide excision of the tumor with resection of adherent structures as much as possbile in order to achieve a tumor-free margin. Radiotherapy and chemotherapy have a limited role in the treatment of retroperitoneal LS. Irradiation therapy improves disease-free survival in extremity LS. However, it is not possible to deliver full-dose radiotherapy to retroperitoneum due to the gastrointestinal and spinal cord toxicity.[20]
Leiomyosarcoma
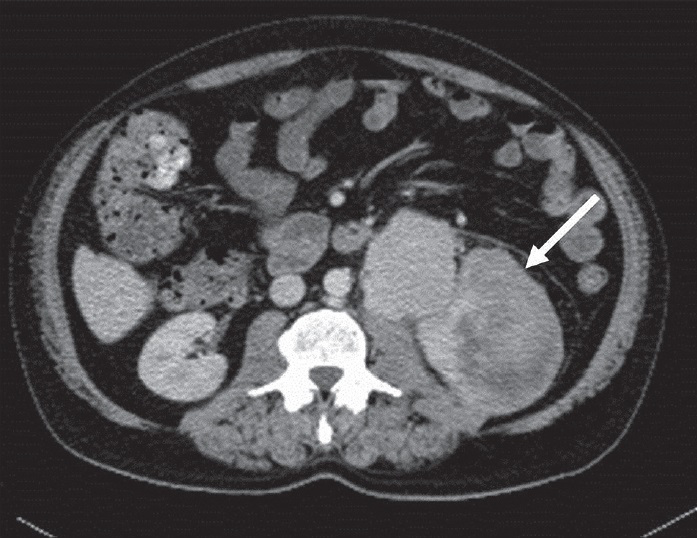
Leiomyosarcoma (LMS) is the second most common primary retroperitoneal sarcoma constituting 28% of all primary retroperitoneal sarcomas.[2] LMS originates from retroperitoneal smooth muscle tissue, blood vessels, or Wolffian duct remnants, and can grow to a larger size and can precipitate clinical symptoms such as venous thrombosis. LMS is more prevalent in women in their 5th to 6th decade of life.[21] While small tumors may appear as a homogeneous solid mass on CT examination, areas of necrosis and hemorrhage may lead to heterogeneity in an LMS. On MRI, LMSs have intermediate-high signal intensity on T2W images and intermediate-low signal intensity on T1W images. Signal intensity can demonstrate variability depending on the amount of necrosis. Small LMSs are usually uniformly solid; however, due to areas of necrosis and hemorrhage, large tumors may have heterogeneous components.[3] Retroperitoneal LMS should be kept in mind if a tumor depicts vascular invasion and extensive necrosis[21] [Figure 9].
- 56-year-old woman with leiomyosarcoma of the left iliac fossa. Post-contrast axial CT image shows a gross enhancing solid mass with extensive necrosis (arrow). Note that the left main iliac artery and vein are infiltrated by the tumoral mass (arrowhead).
IVC involvement is an expected radiologic feature of LMS. The most commonly affected location is the segment between the diaphragm and renal veins. On CT, LMS of the IVC is seen as an intermediate-attenuation mass with heterogeneous enhancement. Intraluminal masses may result in expansion and obstruction of the IVC, and extraluminal masses cause extrinsic compression and proximal dilation. MRI typically shows an intraluminal intermediate-signal-intensity mass with contrast enhancement. Contrast enhancement is the key feature for ruling out benign IVC thrombus.[3]
The cornerstone treatment in LMS is surgery. However, as other sarcomas of the retroperitoneum, locoregional relapse is expected unless complete surgical resection is achieved. Chemotherapeutic agents can also be used in the management of LMS.[22]
Undifferentiated pleomorphic sarcoma
Despite undifferentiated pleomorphic sarcoma (formerly known as malignant fibrous histiocytoma) being the most common soft-tissue sarcoma in whole body, it is the third most common sarcoma in the retroperitoneal space. Fifteen percent of undifferentiated pleomorphic sarcoma occurs in the retroperitoneum and this tumor arises from primitive mesenchymal elements.[2] It is usually seen in the 5th and 6th decades of life and is predominantly seen in men. Undifferentiated pleomorphic sarcoma in retroperitoneal space is a challenging diagnosis radiologically since CT and MRI findings are not spesific. A large, infiltrative, heterogeneously enhancing soft-tissue mass with areas of necrosis and hemorrhage is observed in both CT and MRI [Figure 10]. Invasion of adjacent organs is also expected. A myxoid stroma which shows low signal intensity on T1W, high signal intensity on T2W MR images, and delayed contrast enhancement may be helpful in radiologic distinction. However, except for undifferentiated pleomorphic sarcoma, myxoid stroma may also be seen in myxoid LSs and neurogenic tumors.[3]
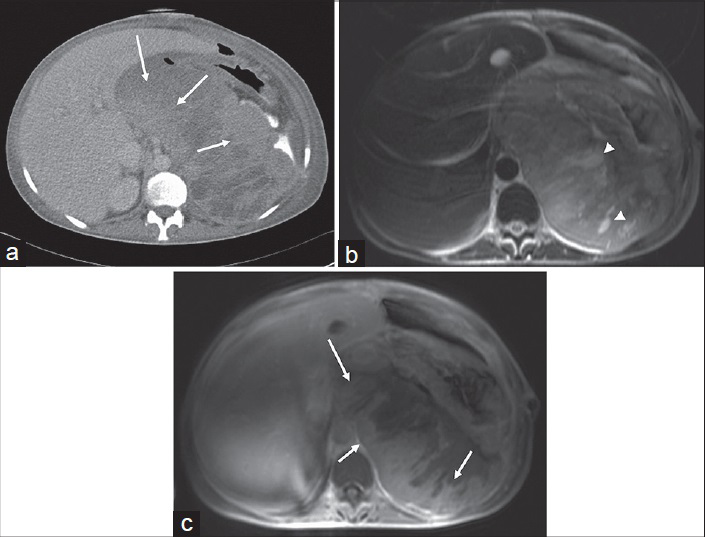
- 39-year-old woman with undifferentiated pleomorphic sarcoma. (a) Post-contrast axial CT, (b) T2W axial, and (c) post-contrast T1W axial images show peripheral heterogeneous contrast enhancement (arrows). T2W axial (b) image demonstrates necrotic portions with high-intensity areas (arrowheads).
The mainstay of the treatment in undifferentiated pleomorphic sarcoma is surgery. The role of chemotherapy and radiotherapy is controversial. Similar to other retroperitoneal sarcomas, local recurrence is often expected because the whole tumor cannot be removed completely.[5]
Rhabdomyosarcoma
Rhabdomyosarcoma, which originates from the primitive mesenchyme, is commonly seen in the pediatric population. The retroperitoneum is involved in 7% of cases diagnosed with rhabdomyosarcoma.[23] In this rare sarcoma of retroperitoneal space, CT and MRI show a mass lesion with areas of necrosis and heterogeneous enhancement [Figure 11]. Calcification is not uncommon.[323]
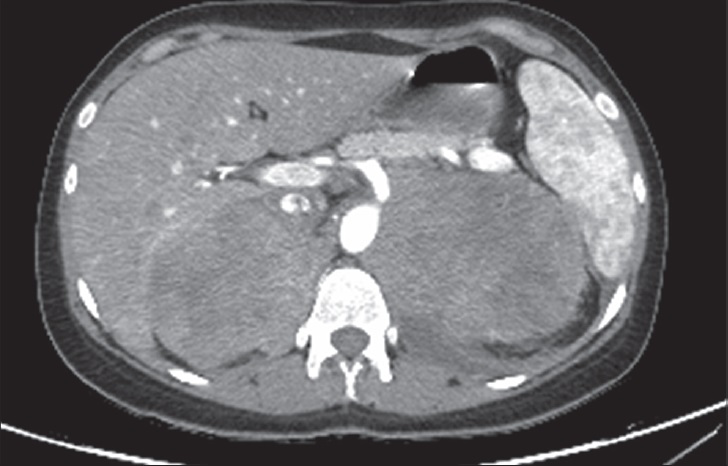
- 34-year-old woman with retroperitoneal rhabdomyosarcoma. Contrast-enhanced axial CT image demonstrates bilateral retroperitoneal extention. The large mass shows heterogeneously enhancing solid portions with patchy necrotic areas.
The standard treatment protocols involve combined therapy including surgery, chemotherapy, and adjuvant radiation.[24]
Extra-gastrointestinal stromal tumor
Extra-gastrointestinal stromal tumors (EGISTs) are neoplasms with histopathologic features resembling those of gastrointestinal stromal tumors (GISTs). However, the distinction of EGIST from GIST is made by proving the origin out of the alimentary system and ruling out a simultaneous neoplasm in the gastrointestinal tract. Retroperitoneal EGIST is a very rare tumor and a total of 58 cases have been reported in the literature.[25] On cross-sectional imaging, retroperitoneal EGISTs are mostly seen as well-defined, inhomogeneous, soft-tissue masses with heterogeneous contrast enhancement. However, they are seldom seen as homogeneous mass lesions[26] [Figure 12].

- 72-year-old woman with extra-gastrointestinal stromal tumor. Post-contrast CT scan shows the tumor as homogeneous and isodense with regard to muscle tissue (arrowheads). Peripheral enhancement is seen in the medial portion (arrow).
Complete surgical removal of the mass and regional lymph nodes is the gold standard therapy for non-metastatic EGISTs. Effective target therapy with oral tyrosine kinase inhibitor (imatinib mesylate) in the medical treatment of EGIST is a controversial issue because of the limited information available in the literature.[25]
NEUROGENIC TUMORS
Schwannoma
Schwannoma is a benign tumor that originates from the perineural sheath of Schwann cell. Schwannoma accounts for 6% of all PRTs. It is more prevalent in women in their 2nd to 5th decade of life.[3] Schwannoma is commonly located in the paravertebral region of the retroperitoneum. Small schwannomas are homogeneous on CT imaging, but large ones may be heterogeneous. Mottled, punctate, or curvilinear calcification may also be present. High cellular areas lead to decreased signal intensity in both T1W and T2W MRI sequences; however, cycstic areas show hyperintensity in T2W images [Figure 13]. Contrast enhancement varies from homogeneous to heterogeneous pattern in both CT and MRI.[27]
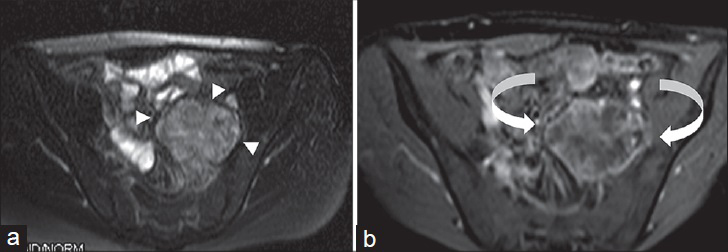
- 24-year-old man dignosed with left pelvic schwannoma. (a) Axial T2W fat-suppressed and (b) post-contrast fat-suppressed T1W images show a heterogeneously hyperintense area in T2W image due to microcytic spaces (arrowheads). Peripheral contrast enhancement is observed in post-contrast T1W image (curved arrows).
Radical excision is considered as the best treatment for retroperitoneal schwannoma. Recent advances such as laparoscopic excision have also been performed in selected cases. Adjuvant chemotherapy and radiation will not add to the benefit of treating schwannomas.[28]
Neurofibroma
Neurofibroma is a benign nerve sheath tumor which occurs either as an isolated tumor or as a component of type 1 neurofibromatosis. It is more prevalent in men in their 2nd to 4th decade of life.[3] Because of lipid-rich Schwann cells, neurofibroma is observed as hypodense round lesions on CT imaging. In contrast to schwannoma, contrast enhancement is more homogeneous. Involvement of the neural foramen gives the characteristic dumbbell shape to the tumor with expansion of the vertebral foramina [Figure 14]. On MRI, neurofibromas are hypointense on T1W sequence and they demonstrate a characteristic target appearance on T2W images with a low signal center surrounded by hyperintense signal caused by myxoid stroma.[27]
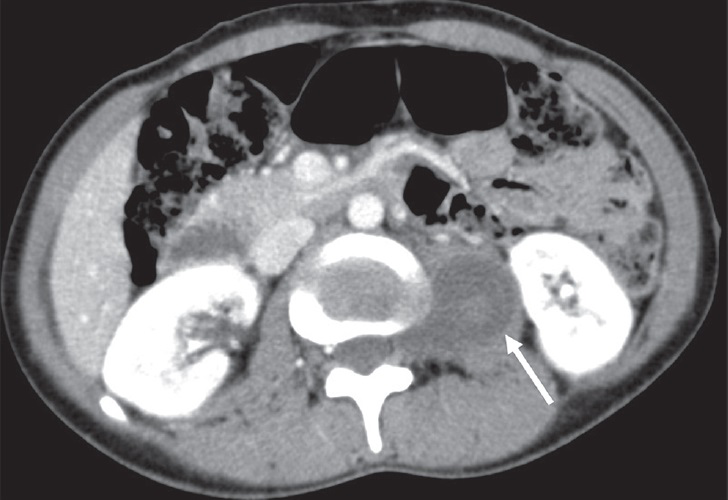
- 28-year-old man with neurofibroma. Axial post-contrast CT image shows a hypoattenuating neurofibroma in the left psoas muscle. Note that left neural foraminal extension is helpful in diagnosis (arrow).
Another form of neurofibroma is plexiform type which is associated with type 1 neurofibromatosis. Plexiform neurofibroma is seen as a large extensive infiltrating mass within the trace of neural fibers [Figure 15]. Malignant degeneration is more common with neurofibroma than with schwannoma, especially in those patients who have neurofibromatosis.[27] Except for the history of neurofibromatosis, myxoid stroma, paravertebral localization, and cystic areas caused by myxoid degeneration are helpful radiologic features that support the diagnosis of neurofibroma.
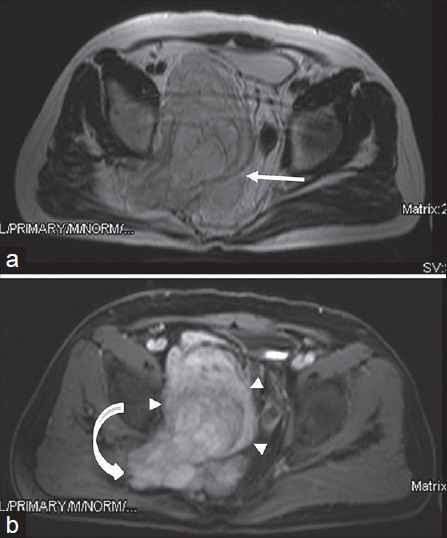
- 73-year-old man with known history of type 1 neurofibromatosis. (a) Axial T2W and (b) T1W fat-suppressed post-contrast images show retroperitoneally located plexiform neurofibroma in the pelvis. Lesion is hyperintense with respect to muscle in T2W image (arrow). Avid contrast enhancement is seen in post-contrast series (arrowheads). Note that plexiform neurofibroma passes through the piriform foramen within the trace of sciatic nerve and gives a dumbbell appearance (curved arrow).
Treatment for neurofibroma basically depends on the symptoms. Since neurofibromatosis cannot be cured, it is feasible to monitor complications and treat neurofibroma-related complaints. Surgical approach performed either with complete or partial resection of the retroperitoneal neurofibroma will help in relieving symptoms.[27]
Paraganglioma
Tumors that arise from the chromaffin cells of the adrenal medulla are called paraganglioma. High catecholamine levels produced by this tumor may lead to sympathetic symptoms such as excessive sweating, hypertension, and, tachycardia. Paraganglioma is usually seen in the 3rd to 4th decades and there is no gender predominance.[3] Paraganglioma can be associated with von Hippel–Lindau syndrome, multiple endocrine neoplasia syndrome, and type 1 neurofibromatosis. Also, mutation in succinate dehydrogenase gene (SDH) may lead to familial paraganglioma. Three subunits of SDH gene mutation have been described in the literature and 33% of patients with SDH subunit B mutation have a positive family history. These aforementioned patients develop single tumors at around 30 years of age and have extra-adrenal paragangliomas mainly in the abdomen and pelvis.[29] Due to the family history and predisposing factors mentioned earlier, if an initial lesion is detected, screening of asymptomatic family members becomes feasible.
On imaging, paragangliomas are often seen as large, well-defined, lobulated mass lesions. Due to hypervascularity, avid contrast enhancement in solid portion is observed. On CT, paraganglioma is usually seen as a large lobular tumor with areas of hemorrhage and necrosis [Figure 16]. Signal voids (due to vascular structures) are seen in T1W images in paraganglioma. Highly hyperintense appearance in T2W images, called “lightbulb,” is characteristic of a classic paraganglioma [Figure 17]. However, due to hemorrhage which leads to heterogeneity, it is sometimes impossible to detect classic “lightbulb” pattern on T2W images.[272930]
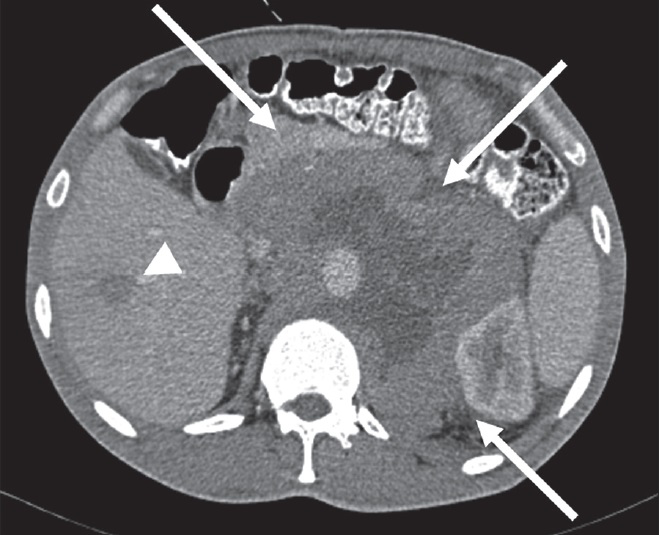
- 42-year-old man diagnosed with malignant paraganglioma. Axial post-contrast CT image demonstrates malignant paraganglioma encircling abdominal aorta in the retroperitoneal region with peripheral enhancement (arrows). Due to extensive necrosis, central portion of the tumor is hypodense with regard to peripheral areas. Note that there is also a liver metastasis (arrowhead) revealing systemic spread.
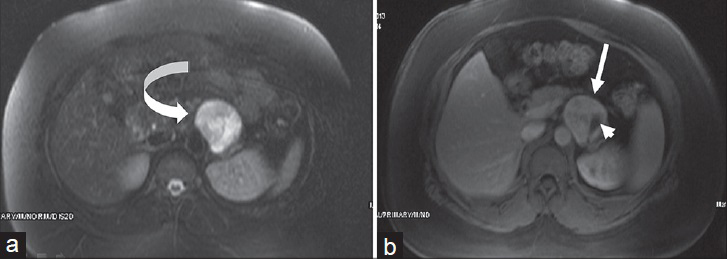
- 39-year-old woman with retroperitoneal paraganglioma. (a) Axial T2W fat-suppressed and (b) post-contrast T1W fat-suppressed images show a left retroperitoneal paraganglioma close to pancreatic corpus. Highly hyperintense appearance is seen in T2W image (curved arrow). Avid contrast enhancement is seen in post-contrast T1W image (arrow). Note that due to vascular structure, a signal void is seen in post-contrast image (arrowhead).
There are several findings that support the diagnosis of paraganglioma. Firstly, hypertension attacks and high blood catecholamine levels obtained from the medical history are in favor of paraganglioma. Secondly, an extremely hypervascular mass in the paravertebral region with fluid–fluid level caused by hemorrhage is highly suggesti ve of paraganglioma.[3]
The traditional treatment of retroperitoneal paraganglioma is complete surgical removal. Because of the malignant potential, monitoring the vascular invasion, liver metastasis, and regional lymph nodes before surgical intervention is critical.[31]
GERM CELL TUMORS
Primary extragonadal germ cell tumors
Germ cell tumors most commonly originate from the testes or ovaries; however, 1–2.5% of germ cell tumors arise in an extragonadal location. After mediastinum, retroperitoneum is the second most common site of extragonadal germ cell tumor (EGCT).[32] Seminomas and nonseminomatous germ cell tumors (yolk sac tumor, embryonal carcinoma, teratoma, mixed germ cell tumors, and choriocarcinoma) are the histologic subtypes of this group. Seminoma in the retroperitoneum is seen as a large, lobulated, well-defined, homogeneous solid mass with peripheral or spotted calcification on CT [Figure 18].[33] Another imaging feature of seminona is fibrous septum, which is hypointense on T2W images and shows enhancement after contrast administration.[333]
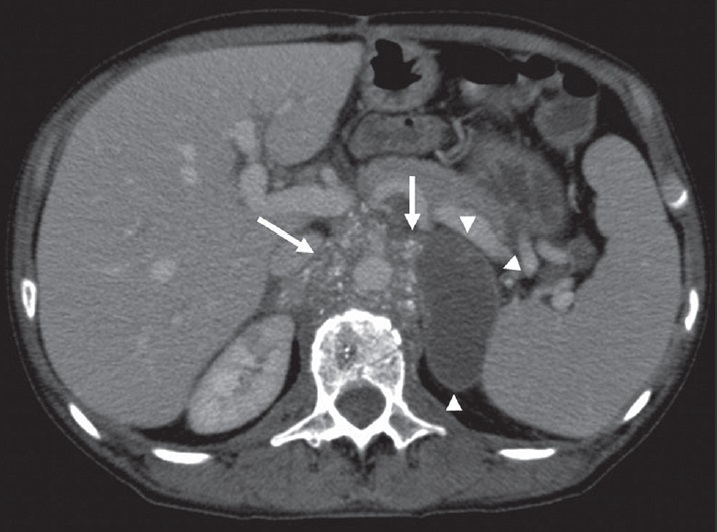
- 64-year-old man diagnosed with extragonadal germ cell tumor. Axial post-contrast CT image of the extragonadal germ cell tumor shows a solid mass encircling aorta with speckled calcification in the para-aortic region (arrows). Tumoral mass lesion contains cystic portion (arrowheads).
Elevation of serum α-fetoprotein levels (particularly in embryonal carcinoma and yolk sac tumor) and increased level of beta-subunit of human chorionic gonadotropin (particularly in choriocarcinoma) are supportive laboratory findings of primary EGCTs. Distinctive radiologic feature of primary EGCTs is that it has a propensity to involve midline structures between T6 and S2 vertebrae.[3]
Chemotherapy and radiotheraphy are the most effective treatment for patients with EGCTs.[34]
LYMPHOID NEOPLASMS
Lymphoma
The most common malignancy in the retroperitoneal cavity is lymphoma, which accounts for one-third of all retroperitoneal tumors.[2] While para-aortic lymph node involvement is seen in 25% of the patients with Hodgkin lymphoma, the percentage is higher in patients with non-Hodgkin lymphoma (55%).[3]
Lymphoma is seen as a well-defined homogeneous mass lesion with homogeneous contrast enhancement. Anterior displacement of aorta and inferior vena cava gives the typical appearance in lymphoma called “floating aorta” or “CT angiogram” sign [Figure 19].[35] After treatment (with chemotherapy or radiotheraphy), calcification and necrosis may be seen in lymphoma.
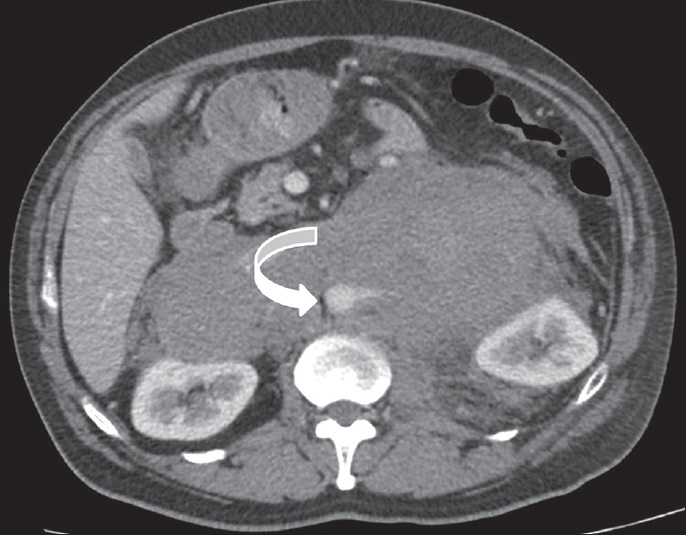
- 37-year-old man diagnosed with non-Hodgkin lymphoma. Axial post-contrast CT image shows a mildly enhancing solid mass surrounding abdominal aorta, giving “CT angiogram” sign (curved arrow).
Lymphoma is usually isointense on T1W images and iso- to hyperintense on T2W images, with moderate homogeneous or patchy enhancement after contrast material administration on MRI. Due to fibrosis resulting from treatment response, contrast enhancement and T2 signal decrease after therapy. Although there are overlaps with other PRTs, lymphoma is seen as a hypovascular mass lesion that spreads between normal structures. Encasement of abdominal aorta and/or IVC without luminal compression is highly suggestive of retroperitoneal lymphoma.
Several chemotherapy regimens are used for treatment of retroperitoneal lymphoma. Chemotherapy, either performed alone or in combination with radiotherapy, is the mainstay of the therapy.[36]
Castleman disease
Castleman disease (CD) is a rare benign condition characterized by lymphocyte proliferation. CD is seen in people of a wide age range (2–85 years), but the median age is 23 years. Women are more affected than men by this disease.[37] CD can be seen anywhere along the lymphatic chain, but the mediastinal lymph nodes are the most common site affected where the disease usually involves, apart from mediastinal lymphatics, axilla, neck, abdominal, pelvic lymphatics, and retroperitoneum.
There are two major histological types: The plasma cell type and the hyaline vascular type. Clinically, the less-common plasma cell variant has a worse prognosis than the latter; thus, it may be disseminated and systemic symptoms (such as anemia, fever, weight loss) are more commonly seen. On the other hand, the hyaline vascular type is usually benign, localized, and asymptomatic.[38]
On CT or MRI, the imaging findings of hyaline vascular type of CD include a well-defined solitary mass or a dominant mass accompanying small satellite nodules, showing intense enhancement. Fibrosis, if seen, is one of the characteristic features, and manifests as hypointense signal on both T1W and T2W images [Figure 20].[38]
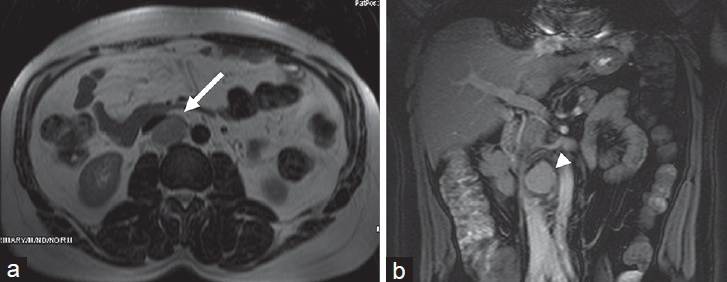
- 53-year-old man with hyaline type Castleman disease. (a) T2W image shows mass lesion is slightly hypointense with regard to fat tissue (arrow). (b) T1W coronal fat-supressed image shows homogeneous contrast enhancement (arrowhead).
Surgical resection remains the standard therapy for localized form of CD; on the other hand, systemic therapies are required for the management of disseminated type of CD. Rituximab, a biologic agent, is the cornerstone of therapy in disseminated type of CD. However, novel therapies targeting IL-6 may represent a treatment option in the near future.[39]
CONCLUSION
A large number of neoplastic entities may arise from the retroperitoneum. Although it is sometimes impossible to make a definitive diagnosis just relying on imaging, cross-sectional imaging in PRTs is indispensable in the pre-operative staging and surgical planning. This article has reviewed the imaging findings and management of different PRTs which can be encountered in daily clinic practice.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2015/5/1/24/156135
Source of Support: Nil
Conflict of Interest: No conflict of interest was declared by the authors.
REFERENCES
- Peritoneal and retroperitoneal anatomy and its relevance for cross-sectional imaging. Radiographics. 2012;32:437-51.
- [Google Scholar]
- CT characteristics of primary retroperitoneal neoplasms. Crit Rev Comput Tomogr. 2004;45:247-70.
- [Google Scholar]
- Primary retroperitoneal neoplasms: CT and MR imaging findings with anatomic and pathologic diagnostic clues. Radiographics. 2003;23:45-57.
- [Google Scholar]
- Retroperitoneal tumours: Review of management. Ann R Coll Surg Engl. 2011;93:275-80.
- [Google Scholar]
- Retroperitoneal cystic masses: CT, clinical, and pathologic findings and literature review. Radiographics. 2004;24:1353-65.
- [Google Scholar]
- CT and MR imaging of extrahepatic fatty masses of the abdomen and pelvis: Techniques, diagnosis, differential diagnosis, and pitfalls. Radiographics. 2005;25:69-85.
- [Google Scholar]
- Differential diagnosis of benign peripheral lipoma from well-differentiated liposarcoma on MR imaging: Is comparison of margins and internal characteristics useful? AJR Am J Roentgenol. 2003;180:1689-94.
- [Google Scholar]
- Fat-containing lesions of the retroperitoneum: Radiologic-pathologic correlation. Radiographics. 2009;29:261-90.
- [Google Scholar]
- Etiology of spontaneous perirenal hemorrhage: A meta-analysis. J Urol. 2002;167:1593-6.
- [Google Scholar]
- Computed tomographic distinction of perirenal liposarcoma from exophytic angiomyolipoma: A feature analysis study. J Comput Assist Tomogr. 2008;32:548-52.
- [Google Scholar]
- Retroperitoneal liposarcoma: MR characteristics and pathological correlative analysis. Abdom Imaging. 2007;32:668-74.
- [Google Scholar]
- Long-term outcomes after radiotherapy for retroperitoneal and deep truncal sarcoma. Int J Radiat Oncol Biol Phys. 2007;69:103-10.
- [Google Scholar]
- From the archives of the AFIP. Leiomyosarcoma of the retroperitoneum and inferior vena cava: Radiologic-pathologic correlation. Radiographics. 1992;12:1203-20.
- [Google Scholar]
- Role of chemotherapy in the management of soft tissue sarcomas. Expert Rev Anticancer Ther. 2010;10:249-60.
- [Google Scholar]
- Imaging and clinical spectrum of rhabdomyosarcoma in children. Clin Imaging. 2000;24:257-62.
- [Google Scholar]
- Spindle cell rhabdomyosarcoma of the retroperitoneum: An unusual case developed in a pregnant woman but obscured by pregnancy. Int J Clin Exp Pathol. 2014;7:4904-12.
- [Google Scholar]
- Primary extra-gastrointestinal stromal tumor of retroperitoneum. Clin Med Insights Oncol. 2012;6:189-97.
- [Google Scholar]
- Retroperitoneal extragastrointestinal stromal tumor: Radiologic pathologic correlation. J Clin Imaging Sci. 2014;4:34.
- [Google Scholar]
- Neurogenic tumors in the abdomen: Tumor types and imaging characteristics. Radiographics. 2003;23:29-43.
- [Google Scholar]
- Endoscope-assisted minilaparotomy (endoscopic minilaparotomy) for retroperitoneal Schwannoma: Experience with three cases. Jpn J Clin Oncol. 2002;32:177-80.
- [Google Scholar]
- MR differentiation of phaeochromocytoma from other adrenal lesions based on qualitative analysis of T2 relaxation times. Clin Radiol. 1997;52:603-6.
- [Google Scholar]
- Extra-adrenal pheochromocytoma: Diagnosis and management. Curr Urol Rep. 2007;8:83-8.
- [Google Scholar]
- Primary extragonadal germ cell tumors of the retroperitoneum: Differentiation of primary and secondary tumors. Radiographics. 1993;13:1365.
- [Google Scholar]
- Extragonadal seminoma presenting as a large mass in the pelvic cavity without c-kit-activating mutations. Jpn J Clin Oncol. 2012;42:650-3.
- [Google Scholar]
- CT “angiogram sign” in primary pulmonary lymphoma. J Comput Assist Tomogr. 1992;16:829-31.
- [Google Scholar]
- Rapid decline of follicular lymphoma-associated chylothorax after low dose radiotherapy to retroperitoneal lymphoma localization. Case Rep Hematol 2014 2014 684689
- [Google Scholar]
- Hyaline-vascular and plasma-cell types of giant lymph node hyperplasia of the mediastinum and other locations. Cancer. 1972;29:670-83.
- [Google Scholar]
- Castleman disease of the abdomen: Imaging spectrum and clinicopathologic correlations. J Comput Assist Tomogr. 2001;25:207-14.
- [Google Scholar]




