
Translate this page into:
Autoimmune Polyglandular Syndrome Type 1
Address for correspondence: Dr. Vedeswari C. Ponranjini, Department of Oral Medicine and Radiology, Sri Ramachandra University, Porur, Chennai, Tamilnadu, India. E-mail: vedeswari80@gmail.com
-
Received: ,
Accepted: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Autoimmune Polyglandular Syndrome (APS) Type 1 is a rare hereditary disorder that damages organs in the body. This disease entity is the result of a mutation in the AIRE gene. It is characterized by three classic clinical features - hypoparathyroidism, Addison's disease, and chronic mucocutaneous candidiasis. For a patient to be diagnosed as having APS Type 1 syndrome at least two of these features needs to be present. The third entity may develop as the disease progresses. We report a case of a 35-year-old female patient with a history of seizure from the age of 11 years, who was managed with anticonvulsant drugs. With worsening of the seizure episodes, patient was diagnosed to have hypoparathyroidism together with the manifestations of oral candidiasis, nails dystrophy, enamel hypoplasia, and hypogonadism. A diagnosis of APS-1 was considered. The facility for genetic analysis of the AIRE gene mutation was not accessible, as the test costs were prohibitive and not affordable for the patient. Patient management was directed to treating individual disease components. However, cerebral and dental changes were irreversible.
Keywords
Autoimmune
candidiasis
ectodermal dystrophy
polyendocrinopathy

INTRODUCTION

Autoimmune Polyglandular Syndrome (APS) are rare endocrinopathies characterized by the coexistence of at least two glandular autoimmune features from a variable combination of (a) failure of parathyroid glands, adrenal cortex, gonads, pancreatic beta cells, gastric parietal cells, and thyroid gland, juvenile onset pernicious anemia and hepatitis; (b) chronic mucocutaneous candidiasis; and (c) dystrophy of dental enamel and nails, alopecia, vitiligo, keratopathy, and calcification of the tympanic membrane.[1] Neufeld et al., in 1980, identified four major subtypes, primarily distinguished according to characteristic patterns of disease combinations. APS Type 1, also known as autoimmune polyendocrinopathy with candidiasis and ectodermal dystrophy or multiple endocrine deficiency autoimmune candidiasis syndrome is an autosomal recessive disorder. It is clinically defined by the presence of at least two conditions of the triad that includes hypoparathyroidism, Addison's disease, and chronic mucocutaneous candidiasis. The order of prevalence of the features increases with age.[2] It develops as a monogenic mutation of autoimmune regulator (AIRE) gene, which maps to 21q 22.3.[3] This mutation affects the normal immunological tolerogenesis leading to the formation of auto-antibodies directed against specific tissue antigens.[2]
CASE REPORT
This case is presented after obtaining the informed consent from the patient. A 35-year-old female under medical care for frequent episodes of seizure was referred to us for evaluation of the symptom of burning sensation in the mouth. History revealed that the patient was a full term normal delivery baby with normal physical and mental development during childhood. At the age of 11 years, she developed seizures that had increased in frequency over the next 6 years. 2 months prior to coming to the hospital, the seizures had become almost a daily occurrence and she was managed with anticonvulsants. She developed bilateral congenital cataracts 10 years earlier and this had gradually progressed to total blindness. She developed primary amenorrhea and lacked secondary sexual characters. General examination revealed her height after the age of 12 years had been consistently below the 5th percentile level, a family history of short stature, flexion contractures of the fingers [Figure 1], and no sensory abnormalities. Dysmorphic nails were evident on the right index finger, left thumb, right great toe, and right fifth toe. The hair on the scalp and eyebrows were normal. The skin was dry with increased facial pigmentation. Intra-oral examination revealed flaring of anterior teeth; clinically missing permanent canines, third molars, right maxillary and left mandibular premolars; retained left maxillary deciduous canine, left mandibular deciduous canine and second molar; and enamel hypoplasia in left mandibular anteriors. Scrappable curdy white patches positive for fungal hyphae were present in multiple sites of the oral mucosa and angular cheilitis was evident [Figure 2].
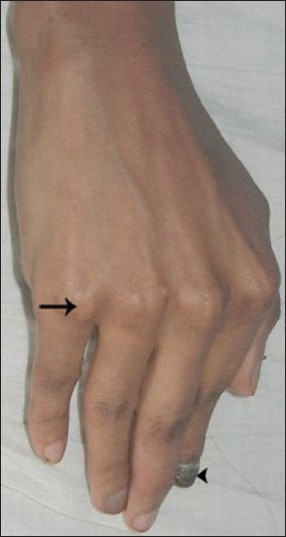
- Right-hand shows flexion contractures of the fingers (arrow) and dysmorphic nail (arrowhead).
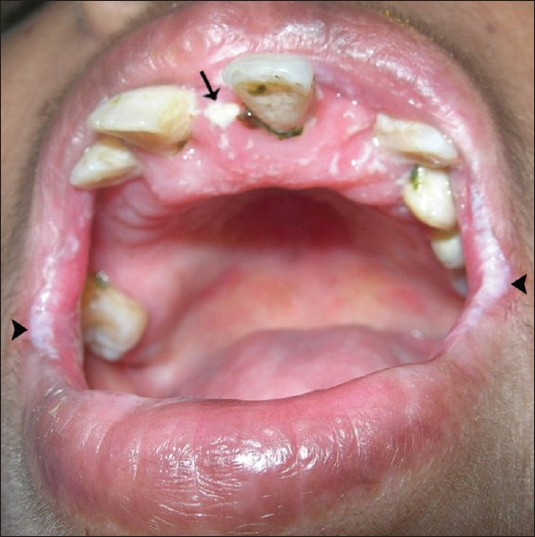
- Intra-oral view of the mucosa shows scrappable curdy white patches (arrow). positive for fungal hyphae and angular cheilitis (arrowhead).
Orthopantomograph [Figure 3] revealed multiple impacted permanent teeth with delayed eruption and incomplete root formation. Laboratory investigations revealed decreased serum calcium 1.25 mmol/L (2.1–2.6 mmol/L) and intact parathormone 0.3 pmol/L (1.2–5.8 pmol/L); elevated phosphorus 2.2 mmol/l (0.8–1.5 mmol/L) and normal magnesium 0.5 mmol/L (0.7–1.0 mmol/L) levels. Other parameters like serum sodium, potassium, cortisol, thyroxine, thyrotropin, plasma glucose, liver function test, and hemogram were within normal limits. Computed tomography (CT) brain [Figures 4, 5] revealed extensive symmetric parenchymal calcifications involving the basal ganglia, cerebellar, and subcorticol regions.
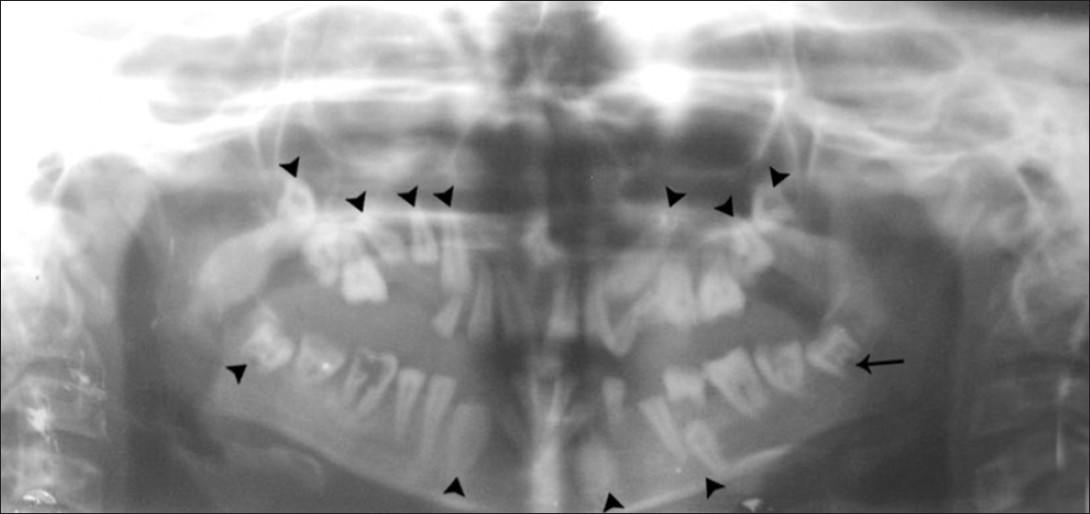
- Orthopantomograph shows multiple impacted permanent teeth (arrow head) with delayed eruption and incomplete root formation (arrow).
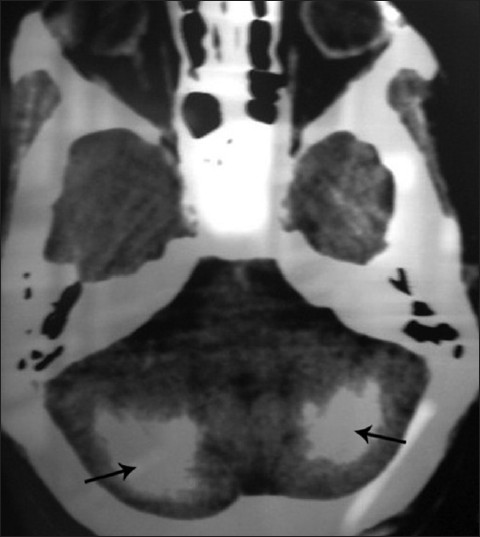
- Axial CT of brain shows diffuse, symmetric parenchymal calcifications in the cerebellar region (arrow).
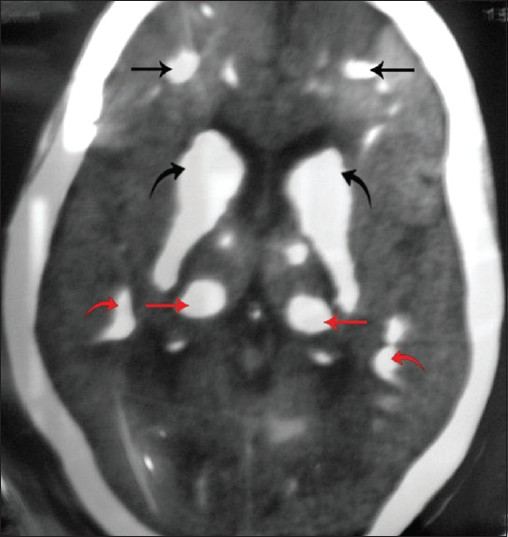
- Axial CT of brain shows extensive symmetric calcifications in the frontal lobe (black straight arrow), periventricular region (black curved arrow), basal ganglia (red straight arrow), and parietal lobe (red curved arrow).
The history and clinical features of hypoparathyroidism, oral candidiasis, nail dystrophy, enamel hypoplasia, and hypogonadism led us to the diagnosis of APS Type 1. Dental management was directed toward symptomatic relief with topical antifungal medication and the patient was referred to an endocrinologist.
DISCUSSION
Although APS-1 is rare, its prevalence is rather high among Finns (1:25,000), Sardinians (1:14,500), and Iranian Jews (1:9,000). The manifestation begins in early life and, therefore, is also called juvenile autoimmune polyendocrinopathy. However, the autoimmune process continues and new complications develop in later years. Earlier the onset, it is more likely that multiple diseases features will develop over time. Our patient presented with hypoparathyroidism and oral candidiasis, the two components of the classic triad of APS-1.[2] The disease components developed during childhood, beginning with bilateral cataracts leading to blindness at the age of 10 years. Seizures started when she was 11 years and later hypoparathyroidism was diagnosed. Oral candidiasis was a more recent development leading to severe burning sensation in the mouth. The patient reported that the symptom had been present for many years but had been milder in intensity and hence had been neglected.
She also presented with hypogonadism, dystrophic nails, and enamel hypoplasia. No evidence of adrenal insufficiency was seen at the time of presentation. The diagnosis of hypoparathyroidism was based on low or undetectable parathormone (<1.6 pmol/L), low calcium (<2 mmol/L), increased phosphorus (>1.6 mmol/L), and normal magnesium (0.7-1.0 mmol/L) levels. Hypoparathyroidism present in majority of cases (80–85%) is often asymptomatic and manifests tetany when calcium levels are significantly reduced. Sometimes, convulsions may be the sole presenting symptom[4] as in our case. Chvostek's sign was negative. The typical age of onset of hypoparathyroidism in APS Type 1 is 5-9 years. The early onset is very likely to reflect autoimmunity. The first seizure episode was at 11 years of age and diagnosis of hypoparathyroidism may have been delayed due to lack of appropriate medical referral and facility.
The patient was treated with Phenytoin for 24 years. Phenytoin can be epileptogenic in the presence of hypocalcemia, as it increases metabolism of active vitamin D to its inactive form by hepatic enzyme induction. Lack of active vitamin D results in reduced calcium absorption from gut and reduced calcium mobilization from bones, leading to worsening of hypocalcemia. This possibility must be considered as the cause of worsening seizure control in our patient who was on Phenytoin, without being supplemented with calcium and vitamin D. Although, chromosome 22q 11.2 deletion syndrome involves both hypoparathyroidism and epilepsy, in the present case there were no typical features of this syndrome including cardiac anomalies or skeletal defects. Chronic hypoparathyroidism leads to visual impairment from cataracts, and if treatment is delayed may lead to total blindness. The progression of congenital cataract can also be enhanced by coexisting hypoparathyroidism, the metabolic disturbance, during which the breakdown of proteins and lipids liberates sulfates, phosphates, and carbonates, which are precipitated by calcium already present in the lens. However, the infectious causes of congenital cataracts were excluded as serology results were negative. Long-standing hypoparathyroidism leads to microscopic colloid deposition around cerebral blood vessels, followed by calcification most commonly in the basal ganglia, thalami, dentate nuclei, cerebral cortex, and mesencephalic gray matter. Patients with hypoparathyroidism may remain asymptomatic or develop Parkinsonian features.[5] The dental findings of delayed teeth eruption and incomplete root formation in idiopathic hypoparathyroidism as reported by Alice Kelly et al.,[6] were observed in our case coinciding with hypocalcaemia during the development of dentition. The enamel defects are mostly hypoplastic in the form of pits, missing enamel, and grooves, correlating with the age of onset of hypocalcaemia. Short roots, hypodontia, and cessation or delay in tooth eruption can also be observed. The oral health of individuals with Autoimmune Polyendocrinopathy-Candidiasis-Ectodermal Dystrophy (APECED) is poor compared with controls and has a higher prevalence of periodontal disease, caries, erosion, and enamel defects.[7]
The next component, mucocutaneous candidiasis is reported to be the initial common presentation[2] occurring in almost 100% of cases, often before the age of 5 years. However, our patient had sought medical advice for symptoms of candidiasis only at 35 years of age. Ahonan et al.[8] reported that candidiasis was not a reliable hallmark of APECED as it was mild, remittent, and appeared much later than the endocrine components. It often affects the oral mucosa and sometimes the esophageal, vulvovaginal, perianal, or intestinal mucosa.[9] The immune dysfunction leads to a selective deficit of T lymphocytes and thus an impaired response to antigens. When the oral mucosa is involved the presentation varies from mild intermittent angular cheilitis to more severe atrophic and hyperplastic forms. Esophageal mucositis with substernal pain and dysphagia, symptomatic intestinal candidiasis with abdominal pain, flatulence, and diarrhoea may occur. Skin and nail infection is limited to no more than 5% of the skin surface.[1] Nails are ridged, discolored, and are often sites of candidiasis.
The other feature of the disorder, Addison's disease was not evident in our patient. This component can manifest prior to, concomitantly with, or following hypoparathyroidism and is often signaled by anorexia, mucocutaneous pigmentation, and weakness. However, it can develop as late as in the fifth decade and hence the patient should be monitored at regular intervals for this feature.[8]
Hypogonadism, presenting as total or partial failure of pubertal development or as premature ovarian failure is more frequent among females (60%) than in males (14%). It appears in 12–60% of the cases.
Ectodermal manifestations[10] other than enamel hypoplasia include dry skin, sparse brittle hair, keratoconjunctivitis, and punctate nail defects. The other less frequent disease components may develop progressively.
Treatment[10] is directed to manage individual symptoms that develop in patients. However, cerebral and dental changes are permanent. The prognosis for APS-1 patients, even if treated, is not good. Regular follow-up with evaluation for organ function helps achieve a better quality of life.
CONCLUSION
APS must be diagnosed in early stages, given its high morbidity and mortality. Treatment should be aimed to preserve the quality of life. Psycho-social support is essential for most patients.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2012/2/1/62/103018
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- Autoimmune polyendocrine syndrome type 1: Case report and review of literature. Arq Bras Endocrinol Metabol. 2012;56:54-66.
- [Google Scholar]
- Reading epilepsy as the initial symptom of idiopathic hypoparathyroidism. Intern Med. 2011;50:1235-7.
- [Google Scholar]
- Calcification of the basal ganglia in chronic hypoparathyroidism. J Clin Endocrinol Metab. 2003;88:1476-7.
- [Google Scholar]
- Cessation of dental development in a child with idiopathic hypoparathyroidism: A 5-year follow-up. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2009;107:673-7.
- [Google Scholar]
- Oral health in Autoimmune Polyendocrinopathy Candidiasis Ectodermal Dystrophy (APECED) Eur Arch Paediatr Dent. 2008;9:236-44.
- [Google Scholar]
- Clinical variation of autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy (APECED) in a series of 68 patients. N Engl J Med. 1990;322:1829-36.
- [Google Scholar]
- Clinical review 93: Autoimmune polyglandular syndrome type I. J Clin Endocrinol Metab. 1998;83:1049-55.
- [Google Scholar]
- APS-I/APECED: The clinical disease and therapy. Endocrinol Metab Clin North Am. 2002;31:295-320.
- [Google Scholar]




