
Translate this page into:
A Case of Mistaken Identity: Glutaric Aciduria Type I Masquerading as Postmeningitic Hydrocephalus
Address for correspondence: Dr. Shabnam Bhandari Grover, E-81, Kalkaji, New Delhi - 110 019, India. E-mail: shabnamgrover@yahoo.com
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
We report the characteristic neuroimaging features of a rare metabolic leukodystrophy in an 8-year-old boy, born of consanguineous parenthood. The child presented with macrocrania, regression of milestones, and dystonia. The patient was referred for magnetic resonance imaging with a clinical diagnosis of postmeningitic hydrocephalus. Imaging revealed ventriculomegaly, diffuse brain atrophy, bilaterally symmetric widened sylvian fissure with temporal lobe hypoplasia, periventricular white-matter hyperintensities, and atrophy with hyperintensity in bilateral basal ganglia was also seen. These imaging features were signatory to arrive at a diagnosis of glutaric aciduria type 1. This disorder may mimic other neurological diseases such as postmeningitic hydrocephalus, which delays the diagnosis. Since early diagnosis and treatment can arrest progression, increased awareness about this condition among radiologists will certainly prevent erroneous diagnosis as had occurred in our patient.
Keywords
Glutaric aciduria type 1
hydrocephalus
macrocrania
magnetic resonance imaging


INTRODUCTION
Glutaric aciduria type 1 (GAT 1) is an autosomal recessive inherited metabolic disease caused by the deficiency of mitochondrial enzyme, glutaryl-coenzyme.[1] It is a rare but important etiology of macrocephaly, with an estimated incidence of 1 in 100,000 births.[2] Past history of febrile illness and presence of macrocrania mimic postmeningitic hydrocephalus. However, classical imaging features lead to the accurate diagnosis of this condition, which is confirmed by biochemical examination of blood and urine. Permanent neurological damage can be prevented by early diagnosis and timely institution of therapy, for which the role of imaging remains unparalleled.
CASE REPORT
An 8-year-old boy was brought with the complaints of progressive cranial enlargement since birth. The parents had a consanguineous marriage with seven offspring and the patient presenting to us was sixth in birth order. There was no significant antenatal or perinatal history. The child had normal milestones till 6 months of age. However, there was a distinct history of an acute episode of meningitis at 6 months of age, following which the milestones regressed. The child could not sit without support and suffered from severe language disabilities, wherein the speech was limited to bi-syllables. The youngest sibling had similar complaints, and all the elder siblings were however normal.
On examination, there was macrocrania and severe muscular hypotonia [Figure 1] with superimposed dystonic movements in the child. Routine laboratory parameters were normal. A provisional diagnosis of postmeningitis hydrocephalus was arrived at by the clinical team, for which the child was referred for magnetic resonance imaging (MRI).
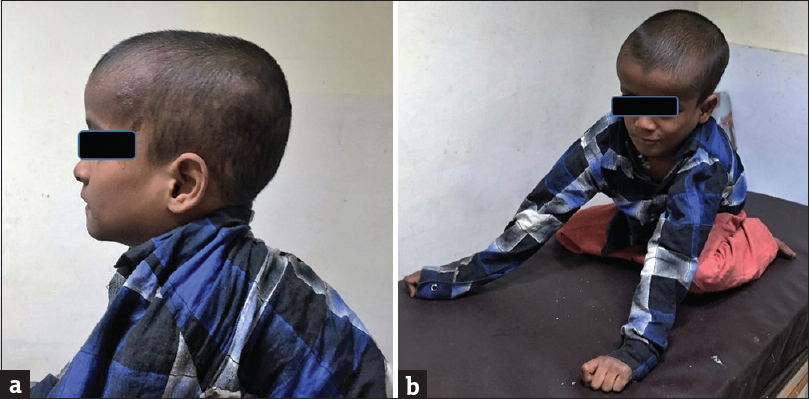
- Clinical images of an 8-year old boy with glutaric aciduria type 1 showing (a) macrocrania, (b) hypotonia and inability of the child to sit without support.
Two prior computed tomography (CT) brain studies had been obtained, one at the age of 15 months and another at 7 years of age at an outside center, which were reviewed by us. Contrast-enhanced CT brain study obtained at 15 months of age revealed mild ventriculomegaly and widened Sylvian fissures. Additionally, there was presence of bilateral nonenhancing hypodense subdural collections along the frontal lobes. However, no CT evidence of enhancing basal exudates was found [Figure 2]. Noncontrast CT study obtained at 7 years of age showed diffuse cerebral atrophy with further enlargement of the ventricular system. Temporal lobe hypoplasia was apparent in the second CT, leading to further widening of CSF spaces anterior to the temporal lobes and widened sylvian fissures. Bilaterally symmetric periventricular hypodensities were also observed [Figure 3]. Both previous CT scans had however been interpreted at the outside center as subdural hygromas, with associated hydrocephalus as a sequelae to meningitis.
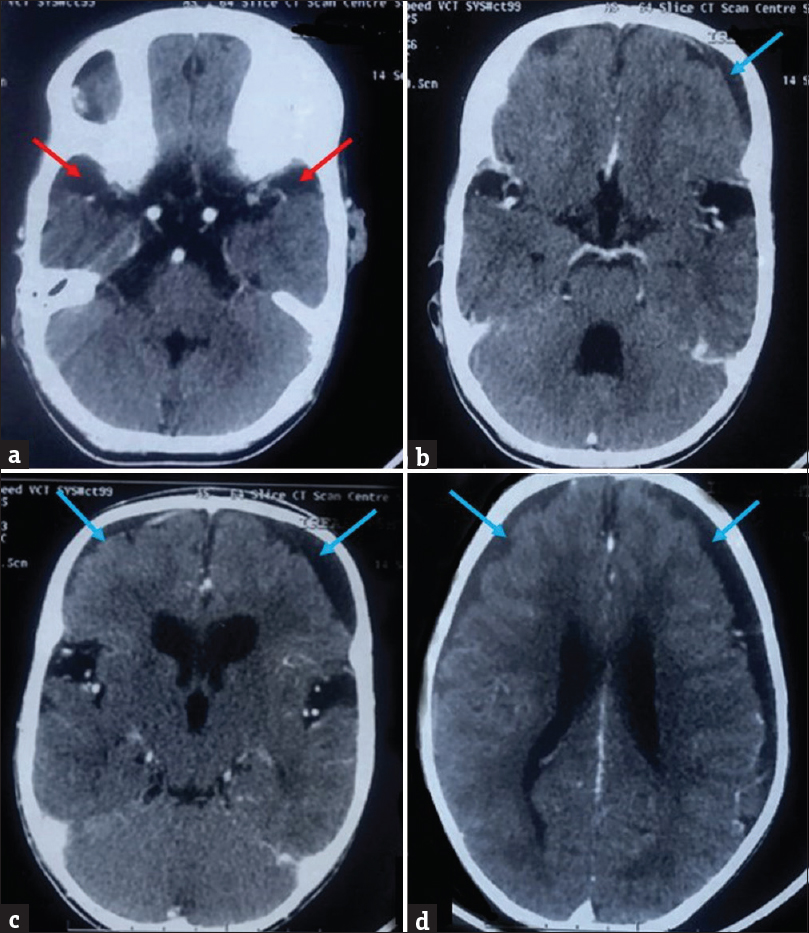
- Prior contrast-enhanced computed tomography brain of the patient with glutaric aciduria type 1, performed at 15 months of age, showing mildly increased cerebrospinal fluid spaces anterior to the temporal lobes (red arrows), prominent sylvian fissures, bilateral subdural collections (blue arrows), and dilated ventricular system. No enhancing basal exudates are noted.
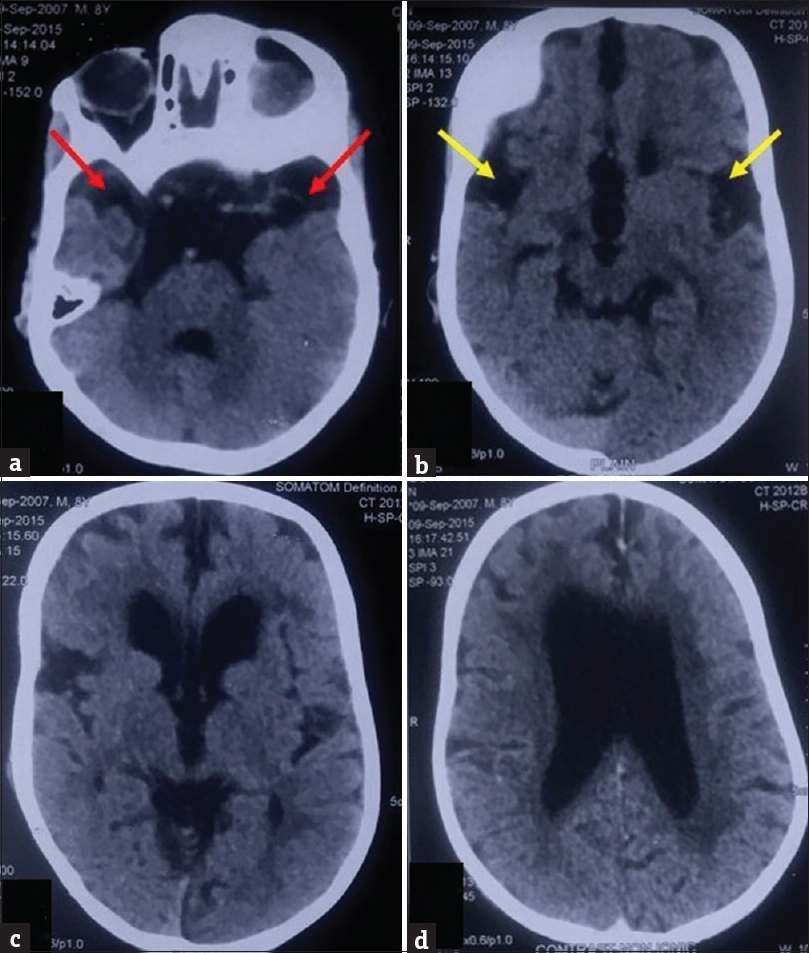
- Prior noncontrast computed tomography (a-d) of the patient with glutaric aciduria type 1, obtained at 7 years of age, showing periventricular white matter hypodensities and cerebral atrophy with predominant involvement of temporal lobes, seen as increased cerebrospinal fluid space anterior to temporal lobes (red arrows) and lack of operculation bilaterally leading to bat wing configuration of sylvian fissures (yellow arrows).
Presently, MR brain study was performed which revealed diffuse brain atrophy. There was symmetric hypoplasia of temporal lobes leading to widening of adjacent CSF spaces. Both Sylvian fissures also appeared widened showing “batwing” appearance. Multifocal confluent periventricular white-matter T2-weighted/fluid-attenuated inversion recovery (T2W/FLAIR) hyperintense signals were also observed. Additionally, there was bilateral symmetric putaminal atrophy with T2W/FLAIR hyperintensities within [Figure 4].
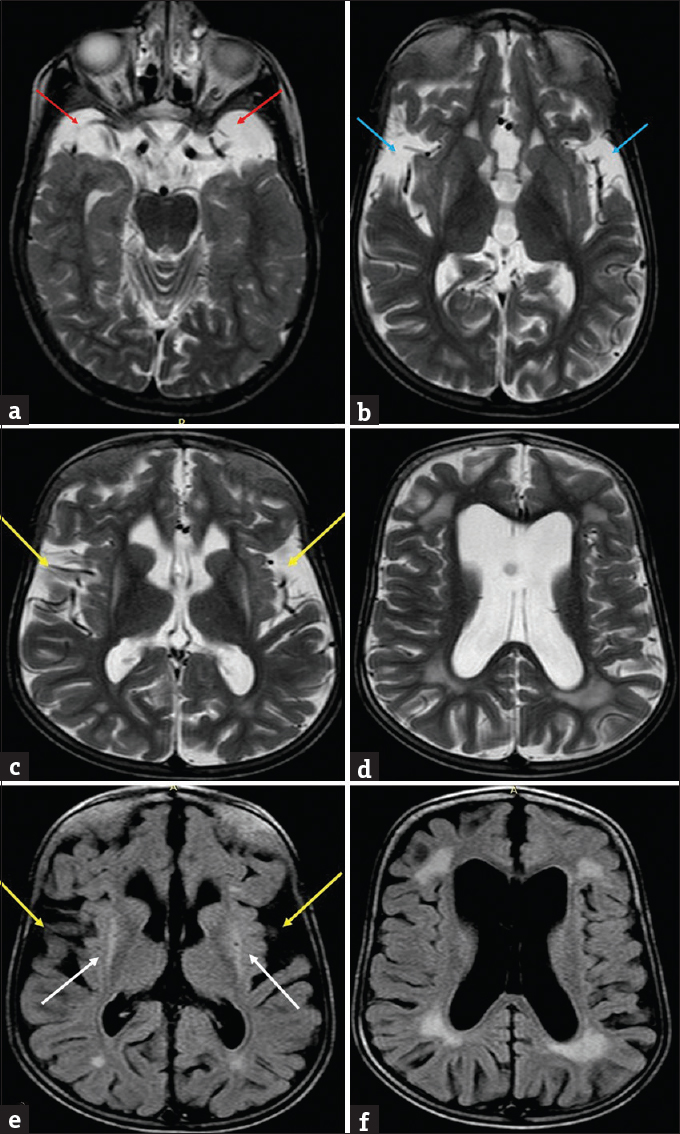
- Present magnetic resonance imaging images of the patient with glutaric aciduria at 8 years. T2-weighted images (a) shows widened cerebrospinal fluid spaces anterior to the temporal lobes (red arrows) with temporal lobe hypoplasia and (b) shows lack of operculation with widened sylvian fissures – “Bat Wing appearance” (blue arrows). Axial T2 (c and d) and fluid-attenuated inversion recovery (e and f) images showing diffuse cerebral atrophy with dilated ventricles, widened sylvian fissures (yellow arrows), periventricular white matter hyperintensities along with hyperintense signal and atrophy of bilateral putamina (white arrows).
These MR features led to the diagnosis of GAT 1, which also explained the appearances on the previous CT studies. Increased levels of glutaric acid in blood and urine were detected with tandem mass spectrometry and gas chromatography/mass spectrometry, respectively, which confirmed the imaging diagnosis.
DISCUSSION
In patients with GAT 1, there is congenital deficiency of the mitochondrial enzyme, glutaryl-coenzyme, which results in abnormal accumulation of glutaric acid in the body. High levels of neural glutaric acid impede cerebral operculation and cause basal ganglia and cerebral white-matter neurotoxicity.[3]
Majority of patients are apparently normal at birth, but present with macrocephaly, regression of developmental milestones, and acute-onset dystonia within the 1st year of life. In most cases, dystonia is triggered by an intercurrent acute febrile childhood infection, usually between 6 and 18 months of age. However, some patients show only an insidious symptomatology presenting with slow-onset hypotonia and choreoathetosis, which gradually progresses into rigidity and dystonia.[34]
The imaging features described for GAT 1 are a characteristic combination of macrocrania, diffuse cerebral atrophy, widened or “open” Sylvian fissures due to lack of operculation (with temporal lobe hypoplasia), diffuse hemispheric white-matter abnormalities, and bilaterally symmetric basal ganglia lesions. These have been described as the “signature imaging signs” for GAT 1.[13] With progression of disease, diffuse cerebral atrophy, ventriculomegaly, and basal ganglia atrophy become more conspicuous. With further chronicity and enlargement of sulcal spaces, there is tearing of bridging cortical veins, resulting in recurrent subdural hematomas. In patients imaged during acute metabolic crisis, there is bilateral edematous basal ganglia which are hyperintense on T2W/FLAIR and show diffusion restriction.[3]
Enlarged ventricular system with subdural collections, seen in GAT 1, can distract both clinicians and radiologists toward postinfective etiology. Central nervous system tuberculosis is usually considered as the culprit, especially in endemic countries. However, the appearances of poor operculation (“batwing Sylvian fissure”), predominant frontotemporal atrophy, and abnormal basal ganglia signal intensities are characteristically present only in GAT 1. Moreover, patients with active tubercular and pyogenic meningitis will have abnormal meningeal enhancement and enhancing exudates in the basal cisterns [Figure 5].
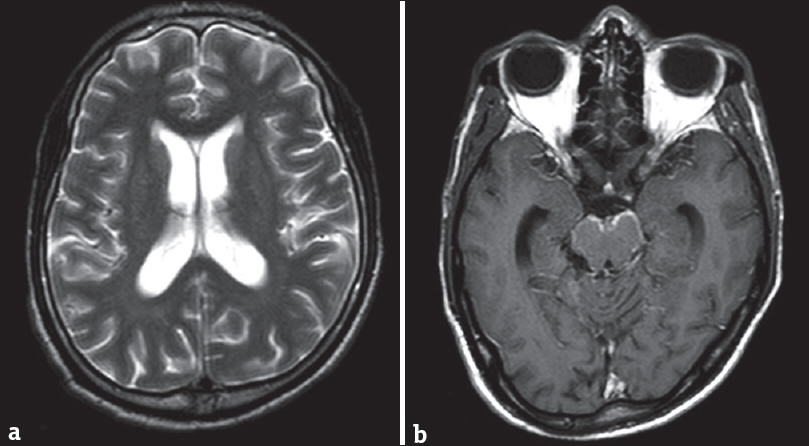
- Magnetic resonance imaging scan of a 15-year old girl with tubercular meningitis. T2 axial scan (a) shows mild communicating hydrocephalous. Contrast-enhanced T1 axial scan (b) shows enhancing basal meninges (b). The sylvian fissures, basal ganglia, and cerebral parenchyma are normal.
An important differential diagnosis of subdural hematomas (SDHs) in children is nonaccidental injuries (NAIs). However, in the absence of associated skull fractures, the diagnosis of NAI can be ruled out. Moreover, in NAI, SDH characteristically extends along the interhemispheric fissure.[5]
Prominent sulcal spaces in infants with macrocephaly can also be seen in the benign expansion of subarachnoid spaces (BESS). BESS is a self-limiting condition with bilaterally symmetric enlargement of subarachnoid spaces in the anterior aspect of the brain with increased interhemispheric and craniocortical distance. It can be differentiated from subdural collections of GAT 1 by the presence of normal cortical veins seen in subarachnoid spaces. Moreover, widened Sylvian fissures and parenchymal signal alterations, seen in GAT 1, will not be seen in BESS.[6] The combination of cerebral atrophy and white-matter hypomyelination can be seen in Zellweger syndrome. However, microgyria, pachygyria, and subependymal germinolytic cysts characteristically seen in Zellweger syndrome are not seen in GAT 1.[3]
Some of the other inherited metabolic disorders causing the combination of macrocephaly with leukodystrophy are Van der Knap disease, Canavan's disease, Alexander's disease, and mucopolysaccharidosis.[3] Van der Knap is a recently recognized leukodystrophy, with diffuse involvement of supratentorial white matter along with characteristic subcortical cysts [Figure 6].[7] In Canavan's disease, MRI shows T2W/FLAIR hyperintensities in the globus pallidus and subcortical white matter, with subsequent involvement of periventricular white matter. On magnetic resonance spectroscopy (MRS), a characteristic N-acetyl aspartate peak is obtained. Alexander's disease shows similar alteration in signal intensities, but predominantly in the bi-frontal white matter, caudate nucleus, and anterior putamina, which characteristically show postcontrast enhancement. Mucopolysaccharidosis may have progressive hydrocephalous and atrophy with macrocrania. However, its classical imaging features include enlarged perivascular spaces with thickened meninges, which are not seen in GAT 1.[3]
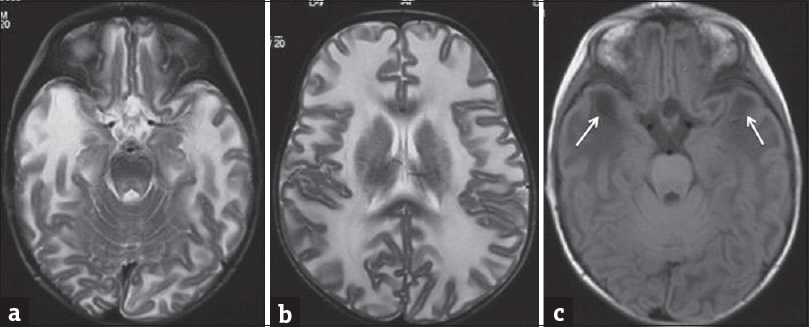
- Magnetic resonance imaging scan of a 1-year-old girl with Van der Knaap disease (megaloencephalic leukodystrophy with subcortical cysts). T2-weighted axial images (a and b) show diffuse white matter hyperintensities with swollen appearance of the brain parenchyma with cyst formation at the temporal poles which is better appreciated on the fluid-attenuated inversion recovery image (arrows in c). (Dr. Atin Kumar, Professor, Department of Radiology, AIIMS, New Delhi, India).
Bilateral T2W/FLAIR hyperintensities in basal ganglia, besides occurring in GAT 1, also occur in Wilson's disease, mitochondrial disorders, organic acidemias, and GAT 2. In Wilson's disease, the ventrolateral aspect of thalamus is involved in addition to the involvement of basal ganglia, cortical/subcortical regions, midbrain, pons, and cerebellum [Figure 7].[8] Leigh's disease is a mitochondrial disorder which shows the involvement of periaqueductal gray matter and cerebral peduncles along with elevated levels of lactate on MRS.[8] Kearns–Sayre syndrome is another mitochondrial disorder in which cerebellum and subcortical white matter are involved in addition to basal ganglia.[3] White-matter signal alterations are observed in both methylmalonic acidemia and propionic academia; however, ventricular enlargement and cortical and cerebellar atrophies occur only in the former. In GAT 2, there is involvement of hemispheric white matter.[3] However, none of the above-described entities show the combination of the signature imaging features of GAT 1. Carbon monoxide poisoning can be effectively excluded by relevant clinical history.[3]
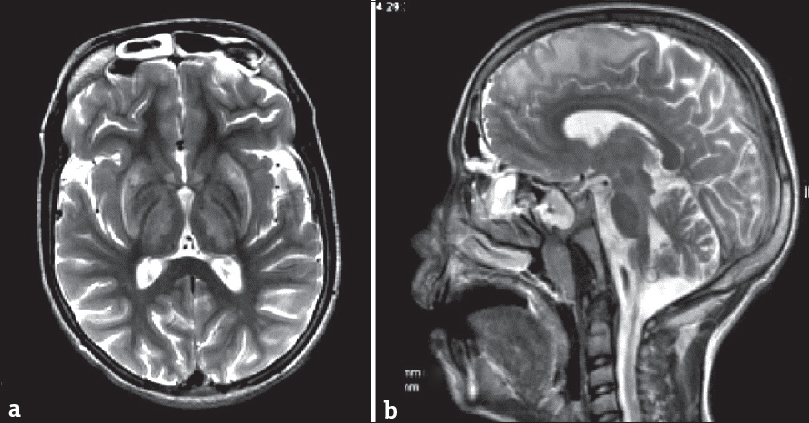
- Magnetic resonance imaging scan of a 14-year-old boy with Wilson's disease. T2-weighted axial image (a) shows bilaterally symmetric hyperintensities in basal ganglia and thalami. Sagittal T2-weighted scan (b) shows hyperintense signal in subcortical white matter in the frontal region and in the midbrain.
The goals of management of GAT 1 are to primarily reduce cellular glutaric acid levels and secondarily institute prompt treatment for intercurrent illnesses. The former can be achieved by dietary restriction of amino acids, which are capable of producing glutaric acid, namely tryptophan and lysine. Additionally, dietary supplementation of carnitine and riboflavin is recommended. Early diagnosis and early therapy reduce the risk of acute dystonia and prevent the progression of the neurological disabilities.[910]
Diligent evaluation with serial neuroimaging studies is an important key not only to clinch the diagnosis, but also to document the progressive nature of GAT 1, as was seen in our patient. Irreversibility of neurological damage highlights the importance of early screening in neonatal period, for metabolic diseases.[2] However, very few countries include the detection of GAT 1 in their routine neonatal screening programs for inborn errors of metabolism.
The radiologist therefore has a critical role in the early imaging diagnosis of this condition as early institution of therapy can halt the progression of the disease. Any diagnostic delay, whether clinical or imaging, leads to progressive and irreversible neurological damage.
CONCLUSION
In underdeveloped countries, the clinical bias toward infectious causes, especially tuberculosis, leads to erroneous interpretation for the etiology of subdural collections and hydrocephalus. Therefore, an astute radiologist plays a vital role in arriving at an early diagnosis of GAT 1, for timely institution of therapy, which can delay the progressive neurological deterioration.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form, the patient's guardian has given his consent for his child's images and other clinical information to be reported in the journal. The patient's guardian understand that his child's names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Acknowledgment
Image Courtesy for Figure 6: Dr Atin Kumar, Professor, Department of Radiology, AIIMS, New Delhi, India.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2018/8/1/50/245531
REFERENCES
- Glutaric aciduria type 1: MR findings in two cases. AJNR Am J Neuroradiol. 1991;12:966-8.
- [Google Scholar]
- Brain MRI findings as an important diagnostic clue in glutaric aciduria type 1. Neuroradiol J. 2013;26:155-61.
- [Google Scholar]
- Inherited metabolic disorders. Osborns's Brain E-Book (2nd ed). Salt Lake City, UT: Elsevier, Inc; 2017. p. :976-1014.
- Nelson Textbook of Pediatrics (20th ed). Philadelphia, PA: Elsevier; 2016.
- From the archives of the AFIP. Child abuse: Radiologic-pathologic correlation. Radiographics. 2003;23:811-45.
- [Google Scholar]
- Benign enlargement of sub-arachnoid spaces in infancy. J Pediatr Neurosci. 2014;9:129-31.
- [Google Scholar]
- Van der knaap disease, a megalencephalic leukoencephalopathy. Indian J Radiol Imaging. 2006;16:733.
- [Google Scholar]
- Differential diagnosis for bilateral abnormalities of the basal ganglia and thalamus. Radiographics. 2011;31:5-30.
- [Google Scholar]
- A treatable neurometabolic disorder: Glutaric aciduria type 1. Case Rep Pediatr 2014 2014:256356.
- [Google Scholar]
- Diagnosis and management of glutaric aciduria type I – Revised recommendations. J Inherit Metab Dis. 2011;34:677-94.
- [Google Scholar]




