
Translate this page into:
Amyotrophic Lateral Sclerosis and its Mimics/Variants: A Comprehensive Review
Address for correspondence: Dr. Vivek S Yedavalli, Department of Neuroradiology and Neurointervention, Stanford University, 300 Pasteur Drive, S047 Palo Alto, California 94305, USA. E-mail: vyedav86@stanford.edu
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Motor neuron diseases (MNDs) are a debilitating subset of diseases, which result in progressive neuronal destruction and eventual loss of voluntary muscular function. These entities are often challenging to distinguish and accurately diagnose given overlapping clinical pictures and overall rarity. This group of diseases has a high morbidity and mortality rate overall and delineating each type of disease can help guide appropriate clinical management and improve quality of life for patients. Of all MNDs, amyotrophic lateral sclerosis (ALS) is by far the most common comprising 80%–90% of cases. However, other mimics and variants of ALS can appear similar both clinically and radiographically. In this review, we delve into the epidemiological, physiological, neuroimaging, and prognostic characteristics and management of ALS and its most common MND mimics/variants. In doing so, we hope to improve accuracy in diagnosis and potential management for this rare group of diseases.
Keywords
Amyotrophic lateral sclerosis
postpolio syndrome
primary lateral sclerosis
progressive bulbar palsy
progressive muscular atrophy
spinal and bulbar muscular atrophy


INTRODUCTION
Motor neuron diseases (MNDs) are a group of diseases which result in progressive destruction of neurons and gradual deterioration of voluntary muscle function, leading to high mortality and morbidity.[12] The average incidence of MNDs is 1–3 cases per 100,000. The prevalence of MND ranges from 1 to 9 cases per 100,000 worldwide. Of this group, amyotrophic lateral sclerosis (ALS) is by far the most common, comprising roughly 80%–90% of MND cases.[3] However, studies have shown at least a 10% error in diagnosis of ALS.[4] ALS and MND, in general, present with nonspecific symptoms such as limb weakness, fasciculations, and fatigue, which can comprise both classic upper/bulbar and lower motor neuron clinical symptoms. In addition, there are no reliable markers to differentiate ALS from other forms of MND, leading to diagnostic dilemmas.
Neuroimaging in conjunction with the clinical picture aids in differentiating subtypes of MND, especially with stratifying upper versus lower MND processes. It is of utmost importance to accurately diagnose and differentiate ALS from its mimics. Delayed diagnosis and treatment can lead to long-term neurological sequelae with far-reaching clinical complications. In this review, our objective is to elucidate the epidemiological, physiological, neuroimaging, and prognostic characteristics of ALS and its most common MND mimics/variants [Table 1].

AMYOTROPHIC LATERAL SCLEROSIS
Epidemiology
ALS, also known as Lou Gehrig's disease, is a degenerative upper and lower MND that leads to progressive weakness, muscle atrophy, fatigue, and eventually death. ALS is a rare disease with an annual incidence of 1.5 per 100,000. Motor neuron diseases, in general, tend to peak in the sixth decade with an overall male predilection. The average lifespan after diagnosis is 3-5 years with respiratory failure as the most common cause of mortality.[5] Most cases of ALS are considered sporadic, but genetic subtypes including autosomal dominant, autosomal recessive, and X-linked recessive patterns have been reported.[36] ALS is a difficult disease to diagnose in a timely fashion due to its nonspecific and varied presentations. Diagnosis is on average delayed up to 12 months from symptom onset with an average of 13.1 months.[7] Delayed diagnosis is more often seen in younger patients, categorized as those under 45.[8]
Pathophysiology
The physiological basis of ALS is not fully understood in the current literature. Genetic factors undoubtedly play a role, but the extent to which has not been fully determined. It is thought that ALS is caused by a prion-like protein dysregulatory process affecting RNA.[9] Other plausible theories include oxidative stress, glutamate excitotoxicity, defects in neurofilament transport, and mitochondrial dysregulation.[6] Loss of Betz cells and aggregation of CD68 macrophages are the pathologic features suggestive of ALS.[10]
Neuroimaging
Neuroimaging plays a crucial role in diagnosis and exclusion of ALS. Conventional magnetic resonance imaging (MRI) is most useful in excluding ALS MND mimics in addition to other spectra of diseases, such as multiple sclerosis or other inflammatory conditions, which can appear radiographically. When suspecting ALS, conventional MR sequences demonstrate bilateral symmetric T2 and FLAIR hyperintensities anywhere along the corticospinal tract superiorly from the cortices extending caudally to the brainstem[11] [Figure 1]. In addition, generalized decreased cerebral volume has been reported, especially in the gray matter of the frontal and temporal lobes.[12] Of note, abnormal FLAIR hyperintensity is nonspecific and does not correlate with disease progression.[13] Within the spinal cord, T1 hypointensity and T2 hyperintensity in the anterolateral portions are characteristic and reflect gliosis and axonal degeneration. Similarly, advanced sequences such T1-weighted magnetic transfer contrast-enhanced imaging show hypointensity in the corticospinal tracts as well.[14] Positron emission tomography (PET) has a valuable role as it demonstrates decreased radiotracer uptake within the frontal and temporal lobes in affected individuals. Verbal fluency can help distinguish ALS on PET from dementia.[15] Advanced imaging techniques such as diffusion tensor imaging (DTI) has shown promise. DTI specifically shows preferential neuronal loss in the primary motor and dorsolateral prefrontal cortices.[16] In addition, it can show decreased fractional anisotropy in the corticospinal tracts, corpus callosum, and thalami, which signify upper motor axonal degeneration.[171819202122] MR spectroscopy (MRS) also has potential value where it demonstrates increased choline and myoinositol metabolic substrates localized to the precentral gyrus.[23] Studies have also shown N-acetyl aspartate/creatine ratios to be abnormal in single voxel evaluation of the corticospinal tracts.[24] These advanced techniques, while promising, currently have low sensitivity and specificity and are not used routinely in clinical practice.[625] A few studies, however, have demonstrated increased utility with DTI/MRS in conjunction, especially on 3T evaluation in diagnosis of ALS and other MNDs.[26]

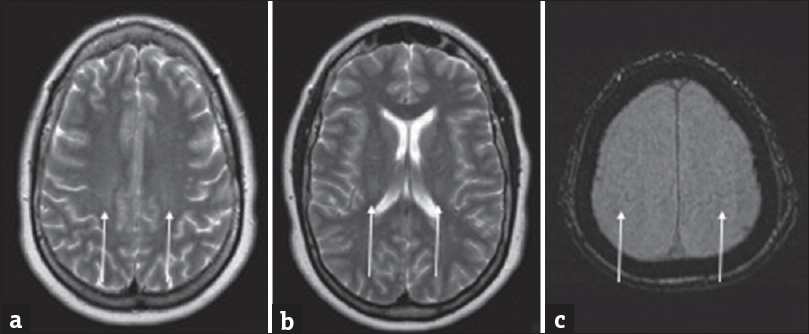
- A 52-year-old male with progressive weakness with findings consistent with amyotrophic lateral sclerosis (ALS). (a-d) Axial T2-weighted images involving the corona radiata (a), posterior limb of the internal capsules (b), cerebral peduncles (c), and midbrain (d).
Clinical presentation and management
Patients afflicted with ALS can present with a number of symptoms, which are mostly bulbar or respiratory in nature. Common bulbar symptoms thought to be due to upper motor neuron (UMN) damage include sialorrhea, facial weakness, gait abnormalities, and dysphagia.[27] Respiratory symptoms can manifest concomitantly or separately, with patients often exhibiting dyspnea, orthopnea, hypoventilation, and respiratory musculature weakness.[27] The generalized weakness, in addition to the aforementioned symptoms, is often gradual and debilitating with progressively worsening muscle wasting, possible cramping, fasciculations, and speech disturbances.[27]
ALS has no cure, but temporary treatments can often provide measures of improved quality of life. Respiratory symptom management is a mainstay of treatment as such complications are the most common cause of death in ALS patients.[27] General treatment options include artificial noninvasive ventilation for dyspnea, orthopnea, and respiratory muscle weakness. Treatment of dysphagia is also crucial, consisting of nutritional supplementation with vitamins, minerals, and creatine in conjunction with enteric feedings if the patient shows weight loss of at least 10% of their prediagnostic weight.[27] Speech therapy has also been beneficial for speech disturbances such as dysarthria.[28] Riluzole, a neuroprotective drug which blocks sodium channels in dorsal root ganglion neurons, has shown promise in treatment of ALS.[2930] Another promising agent that has recently emerged in treatment of ALS is Edaravone. Edaravone is an antioxidant free radical scavenger, which prevents oxidative stress and thereby prevents motor neuron death. In animal models, the medication has been shown to prevent nitration of tyrosine residues within cerebrospinal fluid and improve motor function.[31] Clinical trials have demonstrated some improvement in the revised ALS functional rating scale to assess motor function of patients with Edaravone use as well as slowing of disease progression. Some experts have also advocated combination therapy with Edaravone and Riluzole.[32]
PRIMARY LATERAL SCLEROSIS
Epidemiology
Primary lateral sclerosis (PLS) is a disorder characterized by gradual UMN degeneration, resulting in spinobulbar spasticity with relative sparing of the lower motor neurons.[33] It is typically suggested after at least 3 years of isolated UMN symptoms according to Pringle et al.[3435] The most common clinical symptoms of pure PLS are spasticity, dysarthria, or compulsive laughing/crying.[3436] Studies have shown that spasticity without wasting after at least 3 years is more suggestive of PLS compared to other MNDs.[37] In dedicated MND clinics, PLS is seen in approximately 1%–4% of cases.
PLS subtypes can be distinguished by symptomatic progression or genetic patterns. Symptomatic subtypes include ascending and multifocal, both of which can present asymmetrically.[38] The ascending subtype characteristically shows gradual progression of symptoms from the lower extremities, initially, with subsequent involvement of the upper extremities and, finally, bulbar/pseudobulbar symptoms such as muscle spasticity and dysarthria. Zhai et al demonstrated that upper extremity symptoms on average manifest 3.6 years after the lower extremity presentation with pseudobulbar/bulbar symptoms beginning 1.5 years after the upper extremity symptomatology.[38] The multifocal subtype, on the other hand, shows patchy and asymmetric involvement which can range from initial bulbar symptoms to progressive hemiparesis. Studies have shown that patients who present with initial limb symptoms, amongst the multifocal subtype cohort, have long periods of stability, spanning multiple years, before progression to another limb group.[3839]
In contrast to the symptom related subtypes as described above, PLS can also be characterized based on hereditary patterns, which is labeled as the sporadic subtype. In contrast to the symptomatic subtypes which are often asymmetric in presentation, the sporadic subtype often presents with symmetric paresis. Another distinguishing factor is the scissoring gait presentation, which is also seen in the ascending subtype, but more pronounced with the sporadic form.[39] Irrespective of the subtype, this disease is rare before 29 years of age and is predominantly seen from ages 29-65 with an average onset to symptomatic presentation of 14 years post diagnosis.[39]
Pathophysiology
PLS is classically characterized by dysfunction of the descending corticospinal tracts, resulting in predominantly upper motor disease with degeneration of pyramidal cells in the precentral gyrus, in addition to absence of Betz cells.[40] The exact physiological mechanism is still not fully understood; however, it is theorized to be secondary to mutations in the ALS2 gene, which leads to protein instability and absence of normal function.[41] This is further corroborated with delayed conduction timings on motor evoked potential studies.[40]
Neuroimaging
Neuroimaging can play a critical role in the diagnosis of PLS. PLS is characterized by T2 hyperintensity within the precentral gyrus and adjacent subcortical white matter[42] [Figure 2]. Cortical atrophy primarily within the parieto-occipital regions is also seen.[36] Advanced imaging also proves useful at times with decreased fractional anisotropy and increased mean diffusivity within the corticospinal tracts and mid corpus callosum. These findings are further supported on PET and MRS studies where there is diminished activity in the precentral gyrus and corticospinal tracts.[40]
- A 43-year-old male with excess fatigue compatible with primary lateral sclerosis. (a and b) Axial and sagittal FLAIR images demonstrate faint hyperintensities adjacent to the precentral gyri and along the corticospinal tracts. (c) Axial SWI image with susceptibility signal loss along the precentral gyrus.
Clinical management
Although challenging, PLS has a number of differentiating clinical presentations compared to ALS. PLS is often milder, less diffuse, and more isolated to UMN symptoms only. Patients with PLS tend to be younger at symptom onset.[37] Neurological examination of PLS showing muscle stiffness with the absence of wasting is the key clinical presentation that strongly favors PLS over ALS. PLS also had significantly lower mortality rates over a 16-year period at 33% compared to 89% in ALS.[37] Treatment of PLS is typically tailored in response to specific signs and symptoms of the affected individual. Treatments for spasticity include baclofen, anti-cholinergic medications, or benzodiazepines as well an intrathecal baclofen pump. Physical therapy can also be employed to aid with spasticity. In late disease stages, ventilator support may be necessary for progressive respiratory failure.[43]
Differentiating primary lateral sclerosis from amyotrophic lateral sclerosis
Neuroimaging also plays a supportive role in differentiating PLS from ALS. In conjunction with the clinical symptoms, PLS shows T2 hyperintensity and gradual atrophy isolated to the precentral gyrus and corticospinal tracts secondary to Wallerian degeneration.[3640] In contrast to ALS, PLS often involves the parietal and occipital lobes, while sparing the temporal lobes.[44] While similar, ALS tends to have a more diffuse presentation and rapid onset of these imaging findings. On PET studies, PLS shows hypometabolism in the precentral cortex, while ALS can have frontal lobe involvement, which is not seen in PLS. DTI also can be beneficial where PLS tends to asymmetrically affect white matter tracts while ALS involvement is more diffuse.[40] Fractional anisotropy can also differentiate ALS and PLS as the values are markedly decreased in ALS and not as pronounced in PLS.[45] PLS has a more favorable prognosis compared to ALS.[36] Some studies have shown that persistent isolated upper motor disease without lower motor neuron symptoms at least 3–4 years after diagnosis can result in patients having normal lifespans. As previously stated, PLS overall has a lower mortality rate compared to ALS with a mortality rate of 33% compared to 89% after 16 years of follow-up as shown by Tartaglia et al.[37] Electromyography (EMG) can also be utilized to differentiate the two entities, as EMG is normal or demonstrates minor or transient changes such as sparse fibrillations or fasciculations in the setting of PLS. However, in the setting of ALS, EMG demonstrates widespread motor neuron involvement.[43]
PROGRESSIVE MUSCULAR ATROPHY
Epidemiology
Progressive muscular atrophy (PMA) is an MND characterized by predominant lower motor neuron involvement. It is debated whether PMA is a lower motor neuron subtype within ALS versus a separate entity as certain forms of PMA have significant overlap with ALS. Certain studies favor PMA as a predominantly lower motor neuron presentation of ALS with variable UMN involvement.[1046] However, in the current World Federation of Neurology El Escorial Classification, PMA is viewed as a potential separate entity as “possible” ALS and will be discussed as such in this review.
Pathophysiology
The pathophysiology of PMA has considerable overlap with ALS. Histopathological analysis reveals gradual neuronal degeneration within the pyramidal tracts with ubiquitinated inclusion bodies, which are also seen in ALS.[47] Immunochemical analysis also supports the assertion that PMA is part of the ALS spectrum. DNA binding protein TDP 43 is positive in 85% of PMA and 100% of ALS cases. In addition, studies have shown CD68 macrophage positivity in pathological analysis, once again corroborating the significant overlap between PMA and ALS.[47]
Neuroimaging
MR imaging (MRI) is the mainstay for diagnosis of PMA. The main potentially differentiating imaging factors within PMA are neuronal loss and gliosis within the anterior horns of the spinal cord, manifesting as T2 hyperintensities, in the absence of corticospinal tract involvement [Figure 3]. This is in line with isolated lower MND, which is considered classic PMA.[48] DTI with fractional anisotropy (FA) offers additional utility in diagnosis of PMA in tandem with clinical analysis. In classic PMA, there is absence of corticospinal tract involvement with discrete lower motor neuron involvement.[48] When there is upper MND, DTI demonstrates symmetric and bilateral degeneration of the corticospinal tracts in similar fashion to classic ALS in the UMN and lower motor neuron form. Lower FA values are seen in ALS, PLS, and severe forms of PMA and indicate rapid disease progression.[49]
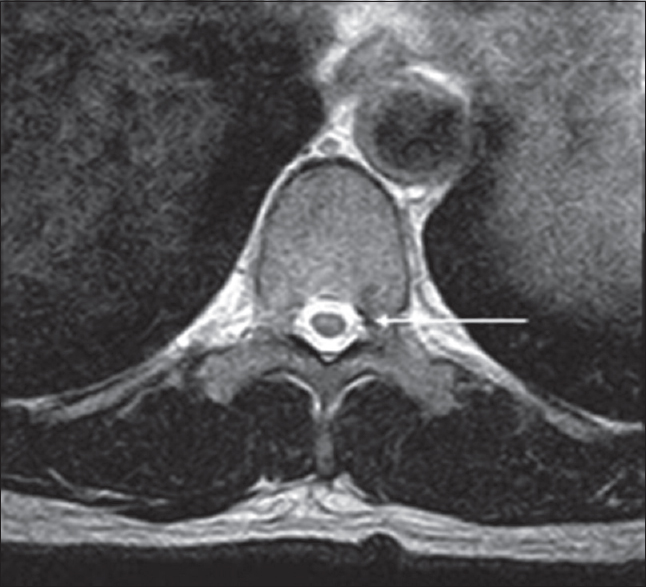
- Adult male with acute-onset weakness compatible with progressive muscular atrophy. There are faint symmetric T2 hyperintensities within the anterior horns of the spinal cord (arrow).
Clinical management
As previously discussed, prognosis in PMA is significantly better than classic cases of ALS with roughly an additional 6–7 years of survival. Treatment, as with ALS, is primarily for symptomatic relief. Given the incidence of respiratory weakness, noninvasive respiratory aids are used to prolong life. Tracheostomies are also performed in severe cases.[50]
Distinguishing progressive muscular atrophy from amyotrophic lateral sclerosis
Although there is significant overlap with ALS, PMA has some distinguishing epidemiological features. PMA has a higher predilection in men and is diagnosed at a later age, with symptoms also presenting at a later age when compared to ALS. In addition, the duration from initial symptomatic presentation to diagnosis is longer in PMA. The most apparent differentiating factor between PMA and ALS is the significantly longer mean survival time of PMA. Patients with PMA survive up to 77 months longer from symptomatic onset when compared to ALS. Distinguishable PMA is seen in approximately 8% of patients.[46] However, isolated lower MND is seen in only 15% of patients with 85% showing both upper and lower MND.[10]
SPINAL AND BULBAR MUSCULAR ATROPHY
Epidemiology
Spinal and bulbar muscular atrophy (SBMA), also known as Kennedy's disease, is a primarily lower motor neuron X-linked recessive disease.[51] The disease tends to occur in individuals aged 18–64 years with most cases in the fourth and fifth decades.[5152] SBMA is rare with an prevalence of 1 in 40,000.[52] Given the genetic component, females with the gene are considered carriers and exhibit no symptoms.[52] The most common clinical symptoms are weakness, tremor, and cramping, often in an asymmetric and bulbar distribution, with associated muscle atrophy. Aspiration and speech dysarthria are also common due to the bulbar musculature being affected.[52] Distinguishing clinical features of SBMA include gynecomastia and diminished fertility in addition to sensory loss in the distal extremities. Laboratory evaluation can be beneficial, generally demonstrating elevated creatine kinase, total testosterone, and free testosterone.[51] Genetic testing is diagnostic.[52]
Pathophysiology
The pathophysiology of SBMA is secondary to a genetic mutation. It is thought to be due to a CAG repeat expansion mutation within an androgen receptor gene on the X-chromosome.[51] CAG repeat mutations are also seen in other entities such as Huntington's disease. This mutation results in a nonfunctional transcribed protein, leading to nonbinding of the testosterone molecule and its derivatives to the receptor.[52] This leads to the commonly identified clinical symptoms and laboratory abnormalities of decreased fertility, gynecomastia, as well as elevated total and free testosterone. Studies have shown that the longer the CAG repeat mutation, the earlier the age of onset for the disease.[53] The specific neurodegenerative pattern is believed to be due to a direct toxic effect from the androgen receptor on both the anterior and posterior horns of the spinal cord in addition to skeletal muscles.[52]
Neuroimaging
MRI is the most useful modality in conjunction with the clinical picture for accurate diagnosis of SBMA. MRI demonstrates atrophy of the anterior and posterior horns of the spinal cord [Figure 4] in addition to the facial, tongue, and respiratory muscles. Involvement of the muscles of mastication is characteristic. DTI has also been utilized, generally demonstrating low FA values in the body and genu of the corpus callosum.[54] Patients require close interval follow-up with video swallow studies to diagnose aspiration.[52]

- Adult male with numbness and weakness in his lower extremities later diagnosed as X-linked spinal and bulbar muscular atrophy. Axial T2 images demonstrate symmetric hyperintensities involving the anterior and posterior horns of the spinal cord (arrow).
Clinical management
Accurate diagnosis and treatment are often delayed due to the initial nonspecific lower motor neuron symptoms. In addition to ALS, this condition is often misdiagnosed for myasthenia gravis (MS). SBMA is generally only suspected after lack of improvement with MS-specific medications in conjunction with negative genetic testing. Although there is no cure, patients generally have normal lifespans as the disease has a gradual onset. Treatment is supportive with a focus on aspiration prevention. Exercise may have potential benefits with mixed results in current literature.[52]
POSTPOLIO SYNDROME
Epidemiology
Poliomyelitis is one of the causes of lower MND secondary to viral infection by the poliovirus, an RNA subtype.[55] With the advent of vaccinations, polio is fortunately one of the many conditions that have been nearly eradicated. However, even in patients who have been afflicted with polio as a child and have recovered, polio can still cause symptoms, resulting in postpolio syndrome (PPS).[55] These patients can present with progressive weakness within the extremities in a lower motor neuron distribution.[56] PPS is defined as new-onset muscle weakness at least 15 years after a fully recovered acute infection.[55] It is clinically characterized by muscle weakness, decreased endurance, pain, and joint deformities.[55] The prevalence of PPS drastically ranges from 15% to 80% depending on the population studied.[57] Some studies have shown a range of 28.5%–64%.[58] Currently, 15–20 million people worldwide are affected by PPS.[55]
Pathophysiology
The etiology of PPS is still debated. The poliovirus initially infects cells of the anterior horns of the spinal cord, resulting in acute paralysis as seen in poliomyelitis.[55] However, PPS is theorized to be Wallerian degeneration, primarily in the anterior horns, of the neuromuscular units, resulting in impairment and muscle atrophy decades after the initial infection.[59] According to such a hypothesis, there is incomplete reinnervation of the affected motor fibers which results in residual weakness and generalized motor impairment that persists for many years.[60] A second hypothesis suggests that PPS is a chronic viral infection. This is supported by findings of oligoclonal IgM bands with poliovirus antibodies in CSF of patients with known PPS. However, the exact mechanism is still widely debated.[55]
Neuroimaging
MRI can corroborate findings of PPS with a concordant clinical picture. MRI shows symmetric T2 hyperintensities within the substantia nigra, pons, medulla, anterior horns of the spinal cord, and ventral nerve roots.[61] In addition to the CNS findings, MRI can also show T1 and T2 hyperintensities within the gluteal musculature without inflammatory changes. These findings are suggestive of diffuse fatty infiltration with concomitant muscular atrophy [Figure 5].[62]
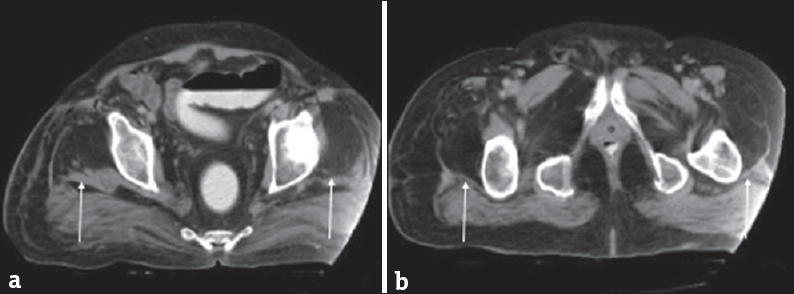
- A 60-year-old man with a history of poliomyelitis now with weakness. (a and b) Axial computed tomographic images demonstrate fatty replacement of the quadriceps musculature (arrows). These findings are seen in postpolio syndrome. Case courtesy of Dr. Ayush Goel, Radiopaedia.org. From the case rID: 34165.
Clinical management
Management of PPS can involve both pharmacological and nonpharmacological therapies. Studies show that treatment with IV Ig therapy in either single or multiple doses is beneficial and results in pain reduction with overall improved quality of life.[55] Dedicated anti-poliovirus medications are also being developed as they target different portions of the virus.[55] Nonpharmacological therapies such as muscle strengthening and rehabilitation are controversial with sparse literature.[55]
PROGRESSIVE BULBAR PALSY
Epidemiology
Progressive bulbar palsy (PBP) is a rapid-onset MND characterized by bulbar symptoms such as difficulty with neck extension, chewing, swallowing, and talking due to weakness of the oral and facial muscles.[63] It can manifest as upper or lower MND and is considered by some to be the bulbar variant of ALS, similar to the lower MND, PMA, which was previously discussed. PBP is seen in 4.1% of MND cases overall, with the highest incidence from ages 51 to 60 years.[64] PBP is more common in elderly women.[56] This disease has a mean survival time of 35–40 months after diagnosis.[65] A rapidly fatal infantile form called Fazio Londe Syndrome (FLS), characterized by cranial nerve palsies and generalized muscle weakness, has also been reported.[66] Case reports also show a similar childhood form, termed Brown-Vialetto-Van Laere Syndrome (BVVL), characterized by sensorineural deafness and ponto-bulbar palsy, resulting in dysarthria and dysphagia.[67] Of note, it is important to distinguish PBP from pseudobulbar palsy, which is a different entity altogether.
Pathophysiology
The pathophysiology of PBP is poorly understood. The childhood forms, FLS and BVVL, are associated with genetic mutations in the SLC52A genes, which encodes a riboflavin transporter and results in a riboflavin deficiency.[68] This can be corrected with riboflavin supplementation.[68] BVVL is also associated with a mutation in the C20orf54 gene. These genetic forms can have both autosomal recessive and/or autosomal dominant inheritance patterns.[69] The adult form of PBP is considered a part of the familial subset of ALS and has been linked previously to mutations in the SOD1 gene, which results in a defective copper/zinc superoxide dismutase protein.[7071]
Neuroimaging
Neuroimaging, particularly MRI, can play a role in diagnosis of PBP. PBP can have nearly identical MRI manifestations to ALS as it is considered by some to be a bulbar variant. In FLS, there are subtle but diffuse subcortical and deep white matter T2 hyperintensities without restricted diffusion.[68] In the adult form, MRI can show T2 hyperintense bands in the frontal lobe, corona radiata, internal capsule, pyramidal tract, and the brainstem [Figure 6]. These findings can appear similar to ALS and are challenging to differentiate.[72]
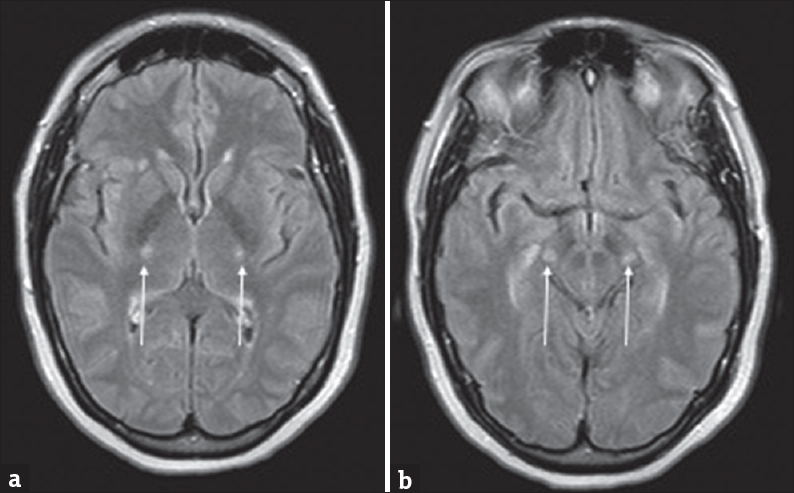
- A 57-year-old female with rapid-onset weakness was favored to represent progressive bulbar palsy. (a and b) Symmetric T2 hyperintensities involving the posterior limbs of the internal capsules and cerebral peduncles of the midbrain (arrows).
Clinical management
Most patients with PBP progress to ALS and prognosis is considered poor as there is no known cure. Mean survival is approximately 40 months after diagnosis.[65] As discussed previously, treatment with riboflavin shows benefits in BVVL.[69] Treatment for the adult form focuses on symptomatic relief of the bulbar symptoms, similar to other MNDs.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2018/8/1/53/247035
REFERENCES
- Neuroepidemiology of amyotrophic lateral sclerosis: Clues to aetiology and pathogenesis. J Neurol Neurosurg Psychiatry. 1996;61:131-7.
- [Google Scholar]
- Amyotrophic lateral sclerosis mimic syndromes: A population-based study. Arch Neurol. 2000;57:109-13.
- [Google Scholar]
- Global epidemiology of amyotrophic lateral sclerosis: A systematic review of the published literature. Neuroepidemiology. 2013;41:118-30.
- [Google Scholar]
- Analysis of the diagnostic pathway and delay in patients with amyotrophic lateral sclerosis in the Valencian Community. Neurología 2018 pii: S0213-4853(18)30157-9. doi: 10.1016/j.nrl.2018.03.026
- [Google Scholar]
- “Aberrant RNA homeostasis in amyotrophic lateral sclerosis: Potential for new therapeutic targets?,”. Neurodegener Dis Manag. 2014;4:417-37.
- [Google Scholar]
- Differential motor neuron involvement in progressive muscular atrophy: A comparative study with amyotrophic lateral sclerosis. BMJ Open. 2014;4:e005213.
- [Google Scholar]
- Amyotrophic lateral sclerosis: Correlation of clinical and MR imaging findings. Radiology. 1995;194:263-70.
- [Google Scholar]
- Whole-brain and regional brain atrophy in amyotrophic lateral sclerosis. AJNR Am J Neuroradiol. 2007;28:255-9.
- [Google Scholar]
- The diagnostic utility of FLAIR imaging in clinically verified amyotrophic lateral sclerosis. J Magn Reson Imaging. 2003;17:521-7.
- [Google Scholar]
- Detection of corticospinal tract compromise in amyotrophic lateral sclerosis with brain MR imaging: Relevance of the T1-weighted spin-echo magnetization transfer contrast sequence. AJNR Am J Neuroradiol. 2004;25:1509-15.
- [Google Scholar]
- Frontotemporal white matter changes in amyotrophic lateral sclerosis. J Neurol. 2005;252:321-31.
- [Google Scholar]
- Diffusion tensor tractography analysis of the corpus callosum fibers in amyotrophic lateral sclerosis. J Clin Neurol. 2014;10:249-56.
- [Google Scholar]
- Topography of the human corpus callosum revisited – Comprehensive fiber tractography using diffusion tensor magnetic resonance imaging. Neuroimage. 2006;32:989-94.
- [Google Scholar]
- Involvement of corpus callosum in amyotrophic lateral sclerosis shown by MRI. Neuroradiology. 1995;37:287-8.
- [Google Scholar]
- The present and the future of neuroimaging in amyotrophic lateral sclerosis. AJNR Am J Neuroradiol. 2010;31:1769-77.
- [Google Scholar]
- Diffusion tensor imaging in amyotrophic lateral sclerosis: Volumetric analysis of the corticospinal tract. AJNR Am J Neuroradiol. 2006;27:1234-8.
- [Google Scholar]
- Diffusion tensor MRI of early upper motor neuron involvement in amyotrophic lateral sclerosis. Brain. 2004;127:340-50.
- [Google Scholar]
- Magnetic resonance imaging in amyotrophic lateral sclerosis. Neurol Res Int 2012 2012:1-9.
- [Google Scholar]
- MR imaging and localized proton spectroscopy of the precentral gyrus in amyotrophic lateral sclerosis. AJNR Am J Neuroradiol. 2000;21:647-58.
- [Google Scholar]
- Motor neuron diseases: Comparison of single-voxel proton MR spectroscopy of the motor cortex with MR imaging of the brain. Radiology. 1999;212:763-9.
- [Google Scholar]
- Diagnostic accuracy of diffusion tensor imaging in amyotrophic lateral sclerosis: A systematic review and individual patient data meta-analysis. Acad Radiol. 2013;20:1099-106.
- [Google Scholar]
- Combined 3T diffusion tensor tractography and 1H-MR spectroscopy in motor neuron disease. AJNR Am J Neuroradiol. 2008;29:1708-14.
- [Google Scholar]
- A comprehensive review of amyotrophic lateral sclerosis. Surg Neurol Int. 2015;6:171.
- [Google Scholar]
- Amyotrophic lateral sclerosis and primary lateral sclerosis: The role of diffusion tensor imaging and other advanced MR-based techniques as objective upper motor neuron markers. Ann N Y Acad Sci. 2005;1064:61-77.
- [Google Scholar]
- Differential action of riluzole on tetrodotoxin-sensitive and tetrodotoxin-resistant sodium channels. J Pharmacol Exp Ther. 1997;282:707-14.
- [Google Scholar]
- Edaravone: A new hope for deadly amyotrophic lateral sclerosis. Drugs Today (Barc). 2018;54:349-60.
- [Google Scholar]
- Clinical efficacy of edaravone for the treatment of amyotrophic lateral sclerosis. Expert Opin Pharmacother. 2017;18:735-8.
- [Google Scholar]
- High T2 signal in primary lateral sclerosis supports the topographic distribution of fibers in the corpus callosum: Assessing disease in the primary motor segment. AJNR Am J Neuroradiol. 2011;32:E61-4.
- [Google Scholar]
- Primary lateral sclerosis. Clinical features, neuropathology and diagnostic criteria. Brain. 1992;115(Pt 2):495-520.
- [Google Scholar]
- Clinical features that distinguish PLS, upper motor neuron-dominant ALS, and typical ALS. Neurology. 2009;72:1948-52.
- [Google Scholar]
- Primary lateral sclerosis: Clinical, neurophysiological, and magnetic resonance findings. J Neurol Neurosurg Psychiatry. 2001;71:615-20.
- [Google Scholar]
- Differentiation between primary lateral sclerosis and amyotrophic lateral sclerosis: Examination of symptoms and signs at disease onset and during follow-up. Arch Neurol. 2007;64:232-6.
- [Google Scholar]
- Primary lateral sclerosis: A heterogeneous disorder composed of different subtypes? Neurology. 2003;60:1258-65.
- [Google Scholar]
- Progression in primary lateral sclerosis: A prospective analysis. Amyotroph Lateral Scler. 2009;10:339-46.
- [Google Scholar]
- “Alsin and the molecular pathways of amyotrophic lateral sclerosis.,”. Mol Neurobiol. 2007;36:224-31.
- [Google Scholar]
- Decreased thickness of primary motor cortex in primary lateral sclerosis. AJNR Am J Neuroradiol. 2007;28:87-91.
- [Google Scholar]
- Primary lateral sclerosis: Clinical and laboratory features in 25 patients. J Clin Neuromuscul Dis. 2005;7:1-9.
- [Google Scholar]
- Serial MRI findings in a case of primary lateral sclerosis. Neurology. 2002;58:647-9.
- [Google Scholar]
- White matter alterations differ in primary lateral sclerosis and amyotrophic lateral sclerosis. Brain. 2011;134:2642-55.
- [Google Scholar]
- Study of 962 patients indicates progressive muscular atrophy is a form of ALS. Neurology. 2009;73:1686-92.
- [Google Scholar]
- “Corticospinal tract degeneration in the progressive muscular atrophy variant of ALS,”. Neurology. 2003;60:1252-8.
- [Google Scholar]
- Diffusion-tensor MR imaging of corticospinal tract in amyotrophic lateral sclerosis and progressive muscular atrophy. Radiology. 2005;237:258-64.
- [Google Scholar]
- Disease course and prognostic factors of progressive muscular atrophy. Arch Neurol. 2007;64:522-8.
- [Google Scholar]
- “Clinical features of spinal and bulbar muscular atrophy,”. Brain. 2009;132:3242-51.
- [Google Scholar]
- “Spinal and bulbar muscular atrophy: Pathogenesis and clinical management,”. Oral Dis. 2014;20:6-9.
- [Google Scholar]
- “Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy,”. Nature. 1991;352:77-9.
- [Google Scholar]
- Brain MR diffusion tensor imaging in kennedy's disease. Neuroradiol J. 2015;28:126-32.
- [Google Scholar]
- “Post-poliomyelitis syndrome as a possible viral disease,”. Int J Infect Dis. 2015;35:107-16.
- [Google Scholar]
- EFNS guideline on diagnosis and management of post-polio syndrome. Report of an EFNS task force. Eur J Neurol. 2006;13:795-801.
- [Google Scholar]
- Inclusion body myositis in post-poliomyelitis muscular atrophy. Acta Neurol Scand. 1988;78:81-4.
- [Google Scholar]
- “Changes in macro electromyography over time in patients with a history of polio: a comparison of 2 muscles”. Arch Phys Med Rehabil. 2004;85:1174-82.
- [Google Scholar]
- MRI findings in children with acute flaccid paralysis and cranial nerve dysfunction occurring during the 2014 enterovirus D68 outbreak. AJNR Am J Neuroradiol. 2015;36:245-50.
- [Google Scholar]
- Progressive bulbar palsy: A case report diagnosed by lingual symptoms. J Oral Pathol Med. 2002;31:277-9.
- [Google Scholar]
- Epidemiological characteristics of motor neuron disease in Chinese patients. Acta Neurol Scand. 2014;130:111-7.
- [Google Scholar]
- The clinical course of progressive bulbar palsy. Amyotroph Lateral Scler. 2010;11:364-8.
- [Google Scholar]
- Brown-Vialetto-Van Laere syndrome, a ponto-bulbar palsy with deafness, is caused by mutations in c20orf54. Am J Hum Genet. 2010;86:485-9.
- [Google Scholar]
- “Early onset of Fazio-Londe syndrome: the first case report from the Arabian Peninsula,”. Hum genome Var. 2017;4:17018.
- [Google Scholar]
- The Brown-Vialetto-Van laere and Fazio Londe syndrome revisited: Natural history, genetics, treatment and future perspectives. Orphanet J Rare Dis. 2012;7:83.
- [Google Scholar]
- SOD1 mutations in amyotrophic lateral sclerosis. Results from a multicenter Italian study. J Neurol. 2005;252:782-8.
- [Google Scholar]
- Unraveling ALS due to SOD1 mutation through the combination of brain and cervical cord MRI. Neurology. 2018;90:e707-16.
- [Google Scholar]




