
Translate this page into:
Clinically Relevant Imaging in Tuberous Sclerosis
Address for correspondence: Dr. Sadhna Verma, University of Cincinnati Medical Center, Department of Radiology, 234 Goodman Street, PO Box 670761, Cincinnati, OH 45267-0761, USA E-mail: drsadhnaverma@gmail.com
-
Received: ,
Accepted: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Tuberous sclerosis (TS), also known as Bourneville disease or Bourneville–Pringle disease, is an autosomal dominant genetic disorder classically characterized by the presence of hamartomatous growths in multiple organs. TS and tuberous sclerosis complex (TSC) are different terms for the same genetic condition. Both terms describe clinical changes due to mutations involving either of the two genes named TSC1 and TSC2, which regulate cell growth. The diagnosis of TSC is established using diagnostic criteria based on clinical and imaging findings. Routine screening and surveillance of patients with TSC is needed to determine the presence and extent of organ involvement, especially the brain, kidneys, and lungs, and identify the development of associated complications. As the treatment is organ specific, imaging plays a crucial role in the management of patients with TSC.
Keywords
Angiomyolipomas
lymphangioleiomyomatosis
tubers

INTRODUCTION

Tuberous sclerosis complex (TSC) is a rare autosomal dominant neurocutaneous syndrome characterized by the presence of benign congenital tumors in multiple organs. It has a birth incidence of 1:6000, with over two-thirds of cases being sporadic from new mutations. TSC can affect virtually any organ system[1–3] and all racial groups. Hamartomas in TSC patients are frequently present in the skin, brain, kidneys, and heart. Less frequently, hamartomas involve the lungs, retina, gingiva, bones, and gastrointestinal tract.[4]
TSC most often presents with neurologic symptoms, with approximately 90% of affected individuals experiencing seizures and about half the patients experiencing cognitive impairment, autism, or other behavioral disorders. Neurologic symptoms are generally due to hamartomas. Renal manifestations are the second most common finding associated with TSC, with angiomyolipomas (AMLs) occurring in 80%, and renal cystic disease in 50% of the patients. Pulmonary involvement, specifically lymphangioleiomyomatosis (LAM), is the third most common cause of TSC associated morbidity, occurring in approximately 35% of female TSC patients.
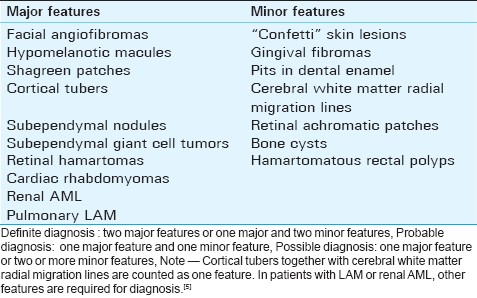
The classic Vogt's triad (facial angiofibromas, mental retardation, and intractable epilepsy) is present in less than 40% of affected patients. In a consensus conference in 1998, Roach et al[5] revised the clinical diagnostic criteria of TSC to provide a standardized approach for identification of this disease. There are many features that are commonly present in TSC, but not necessarily diagnostic in isolation, as they may also be seen in a small percentage of the normal population, thus necessitating a standardized approach. Features that were more specific for TSC formed the major features and features that were less specific for TSC were included in the minor features. Both objective clinical signs and imaging findings were included. Some features that were occasionally seen together in the unaffected population (such as cortical tubers and white matter migration lines) were considered a single criterion if present together in TSC [Table 1]. The presence of typical imaging features of TSC may help to confirm diagnosis in patients with suspected TSC.
GENETIC BASIS
TSC is caused by mutations in either of the two tumor suppressor genes known as TSC1 and TSC2 that have been mapped by linkage analysis in multigenerational families and positional cloning.[67] The TSC1 gene consists of 23 exons located on the long arm of chromosome 9 (9q34) and encodes a 130-kDa protein called hamartin. The TSC2 gene consists of 41 exons on the short arm of chromosome 16 (16p13) and encodes a 200-kDa protein called tuberin. The location of the TSC2 gene is contiguous with the PKD1 gene, which is involved in polycystic kidney disease, and this proximity may explain the presence of multiple renal cysts in patients with TSC.[8] More than 200 TSC1 and almost 700 TSC2 allelic variants have been reported.[910] Pathogenic mutations can be identified in up to 85% of TSC patients using current genetic tests.[11]
Hamartin and tuberin interact with high affinity to form a complex in multiple organs, such as the kidneys, brain, lungs, and pancreas. In these organs, the hamartin-tuberin complex plays a critical role in the integration of cellular sensory input including cellular energy supply, growth factors, genomic integrity, and growth substrate availability. Advances in cytogenetics and pathophysiology have brought to light the functions of the hamartin-tuberin heterodimer. Normally, tuberin is inactivated by direct phosphorylation, resulting in downregulation of the Ras homologue expressed in brain (Rheb), a specific GTPase that in turn blocks the activity of the main mammalian regulator of cell growth and proliferation, mTOR kinase. Hamartin is also believed to influence mTOR activation.[1] If either tuberin or hamartin is absent, as in TSC, mTOR is upregulated, enabling cell growth and proliferation.[12] Loss of heterozygosity of the TSC1 or TSC2 gene is identified in most renal AMLs, subependymal giant cell astrocytomas (SEGAs) in the brain and cardiac rhabdomyomas.[1314]
CNS LESIONS
The common brain lesions encountered in TSC include cortical and subcortical tubers, subependymal nodules (SENs), SEGAs, and white matter lesions.[1516] Estimated prevalence of cortical tuber and/or SENs is 95–100% and that of white matter abnormalities is 40–90%.[17–20] Cortical tubers and SENs have been diagnosed in utero with fetal magnetic resonance imaging (MRI) as early as the second trimester.[21] In addition to the classic intracranial lesions in TSC, developmental dysplasias of the brain are described that are less commonly identified. Cortical dysplasia may range from focal gyriform abnormalities and heterotopias to more extensive lobar migration abnormalities such as schizencephaly. Large segments of cortical dysplasia involving entire lobes or a hemisphere, termed transmantle cortical dysplasia, may be present, with characteristic calcifications when associated with TSC.[22] Partial agenesis of the corpus callosum has been described in TSC. There are several case reports of intracranial aneurysms, frequently in the anterior circulation. Intracranial moyamoya vasculopathy has also been described as a consequence of carotid artery disease in a few patients. Ventricular enlargement may be seen secondary to hydrocephalus or due to volume loss in dysplastic brain.[2324] Infratentorial lesions are less commonly identified in patients with TSC. Apart from tubers, SENs and white matter lesions, less frequent infratentorial lesions include cerebellar dysplasia, cerebellar folia or white matter calcifications, cerebellar hyperplasia, and chiari malformation.[2324]
Cortical tubers
Cortical tubers are areas of markedly disorganized cortex with loss of the normal six-layer architecture of the cortex. These regions contain aggregates of abnormal glial and dysplastic neuronal elements including giant cells or balloon cells, which are characteristic of tubers. Cortical tubers are mostly supratentorial, in the frontal lobes, although infratentorial cerebellar tubers have been described as well.[25] The number and location of these tubers is associated with the degree of cerebral dysfunction including cognitive impairment, seizures, and autism.[26–28] There have been attempts to describe clinical severity based on MRI imaging characteristics [T1, T2, and fluid attenuated inversion recovery (FLAIR) signal] of cortical tubers,[29] but larger studies are needed to establish reliable criteria.
MRI is the most sensitive modality in the identification of cortical tubers that are commonly located at the gray-white junction, but frequently involve the overlying cortex and the subcortical white matter. Often, the cortex overlying a tuber is dysplastic and may demonstrate pachygyria or polymicrogyria. In older children and adults, the tubers have low signal on T1-weighted images and are hyperintense on T2-weighted and FLAIR images. In neonates and infants, with a background of unmyelinated white matter, cortical tubers have high signal on T1-weighted images and low signal on T2-weighted images. Due to variable T2 white matter signal in neonates, cortical tubers can be difficult to detect on T2-weighted imaging. Tubers frequently demonstrate central cavitation or calcification (50%), and approximately 10% of tubers demonstrate enhancement following contrast administration [Figure 1].

- A 21-year-old woman with tuberous sclerosis. (a) Axial T2-weighted images of the brain at multiple levels. There is a dominant hyperintense T2 lesion in the left temporal cortex and subcortical region consistent with cortical tuber (white arrows). (b) Cortical tubers are also seen in the right insula and right parieto-occipital region (white arrows). There is a prominent subependymal nodule at the foramen of Monro (arrowhead). (c) Smaller subependymal nodules are identified along the walls of the lateral ventricle (black arrows).
Occasionally, cortical tubers act as epileptogenic foci and may be treated with surgical resection if antiepileptic medication is ineffective. MRI, electroencephalography (EEG), and nuclear medicine imaging techniques such as positron emission tomography (PET) and single-photon emission computed tomography (SPECT) are used to localize the epileptogenic focus before surgery [Figure 2]. Ictal SPECT imaging can demonstrate increased radiotracer activity corresponding to increased perfusion in the epileptogenic region.

- A 9-month-old boy with tuberous sclerosis. (a) Color-coded axial PET image through the brain shows a focus of decreased radiotracer activity in the left frontal convexity. (b) Axial T2-weighted MRI through a comparable location demonstrates multiple hyperintense cortical and subcortical lesions, one of which corresponds to the hypometabolic focus on PET. (c) Axial ictal brain perfusion scan shows persistent decreased activity in the region of tuber in the left cerebral cortex (arrowhead), with increased radiotracer activity in the right frontal region indicating an epileptogenic focus (arrow).
Subependymal nodules
SENs are present in about 80% of TS patients and are located around the wall of the lateral and third ventricle, usually in close proximity to the foramen of Monro.[25] Histologically, SENs contain large cells, similar to giant cells seen in tubers and elongated glial cells and are covered by a thin layer of ependyma. On MR imaging, these lesions are isointense or hyperintense on T2-weighted sequences and hyperintense on T1-weighted sequences. SENs may also demonstrate central low T2 signal with surrounding hyperintense T2 rim.[19] Variable enhancement may be demonstrated following contrast administration. SENs are more prone to calcification than cortical tubers, particularly at an early age, and are readily detected by computed tomography (CT) or susceptibility weighted imaging [Figure 3]. SENs typically measure less than 1 cm in size, but may grow over time and give rise to SEGAs.

- A 20-year-old woman with tuberous sclerosis. (a) Axial FLAIR MR shows small subependymal nodules along the lateral walls of the lateral ventricles (white arrows) and heterogeneous masses at the foramen of Monro that likely represent subependymal giant cell astrocytomas (arrowheads). There is cortical and sub-cortical bright signal indicating tubers (black arrows). (b) Noncontrast head CT at the corresponding location shows calcifications at the subependymal giant cell astrocytomas (arrowheads), subependymal nodules (white arrows) and cortical tuber (black arrows) on the right.
Subependymal giant cell astrocytomas
SEGAs, although rare, are the most common brain tumors, occurring in up to 26% of TSC patients. The peak age of SEGA is during adolescence, although they have also been identified in utero. Microscopically, SEGAs consist of a mixed cell population of dysmorphic glial cells and giant cells with a rich vascular stroma. They are WHO grade 1 tumors with slow growth, minimal parenchymal invasion, absence of adjacent brain edema and no predilection for CSF spread. It is widely accepted that SEGAs arise from SENs, and serial CTs demonstrating growth of SEN into SEGA have been described [Figure 4].[30] Intermediate cells are identified between hamartomatous SEN and SEGA.[30] SEGAs commonly are located at the foramen of Monro, and their rapid growth can result in rather acute obstructive hydrocephalus. SENs near foramen of Monro, that increase in size over time, should be viewed as suspicious for SEGA. SEGAs are heterogeneous on MR imaging (T1 iso/hypointense, T2/FLAIR iso/hyperintense), contain calcifications, and demonstrate intense inhomogeneous enhancement. With the development of non-communicating hydrocephalus, transependymal flow of CSF may be apparent. In view of their vascular nature, CT and MRI may be useful in noninvasive assessment prior to surgery and in yearly screening during the high-risk period (8–18 years). MR spectroscopy may be useful in distinguishing SEGAs with high Cho/Cr and low NAA/Cr ratios from SENs.[31] In the absence of stigmata of TSC, a mass at the foramen of Monro may bring forth a differential diagnosis of choroid plexus papilloma, meningioma or neurocytoma. Choroid plexus papillomas are more common in infants than in adults, where they typically are located in the atrium of the lateral ventricle. Neurocytomas are usually attached to the septum pellucidum, and meningiomas, although rare in this location, are typically attached to the choroid plexus when intraventricular.

- Sequential gadolinium-enhanced, axial T1-weighted images through the brain. (a) At 12 years of age, the image demonstrates a small, enhancing subdpendymal nodule near the foramen of Monro on the right. (b) At 20 years of age, there is a large, enhancing lobulated lesion in this location which developed in a span of 8 years, consistent with a subependymal giant cell astrocytoma. (c) At 21 years, there is mild decrease in the size of subependymal giant cell astrocytoma following initiation of rapamycin.
SEGAs are usually followed by MRI for changes in size and onset of hydrocephalus, which may necessitate CSF diversion procedures, such as placement of a ventriculoperitoneal shunt. Surgical resection of SEGA is curative with good outcome and a low recurrence rate. Currently, surgery is planned before the onset of obstructive hydrocephalus. Sirolimus and everolimus are therapeutic agents, and both act by inhibiting mTOR (see above); they are used for management of SEGA in nonsurgical candidates. Everolimus has recently been approved by the FDA (in 2010) for treating SEGA.
White matter lesions
Superficial white matter abnormalities, radial white matter bands, and white matter cystic lesions occur in TSC. While white matter abnormalities can occur in all parts of the brain, they are most frequent in the frontal and parietal lobes and occasionally occur in the cerebellum.[19] Superficial white matter abnormalities are associated with almost all cortical tubers and exhibit T2 hyperintensity reflecting gliosis or decreased myelin content. Radial white matter bands are radiating lines of abnormal bright T2 and FLAIR signal extending from the periventricular regions into the subcortical white matter and are believed to represent lines of arrested neuronal migration. In the incompletely myelinated brain, white matter lesions are hyperintense to unmyelinated white matter on T1-weighted sequences and iso to hypointense to the white matter on T2-weighted sequences[19] [Figure 5]. Cysts are present in approximately 44% of patients with TSC and are frequently smaller than 1 cm and located adjacent to the occipital horn or trigone of the lateral ventricle. These cysts follow CSF signal on all sequences and may be septated or demonstrate peripheral enhancement. The etiology of these cysts is unclear.[18]

- A 4-month-old infant with growth retardation. (a) Axial FLAIR image demonstrates a subependymal nodule at the foramen of Monro (arrowheads), but no definite white matter changes. (b) Corresponding T1-weighted image better demonstrates the white matter and cortical lesions (arrows) in this infant with incomplete myelination
RENAL INVOLVEMENT
Renal manifestations are the second most common findings associated with TSC. Both renal cystic disease and AMLs cause chronic renal disease, affecting approximately 1 million patients with TSC worldwide. Using death certificate data, Shepherd et al identified renal failure as the leading cause of death in adult patients with TSC at the Mayo Clinic.[32]
Renal angiomyolipomas
Benign AMLs are the most common renal lesions in adults and older children with TSC.[133] In comparison to sporadic AMLs, renal AMLs in TSC develop in early childhood, grow throughout adolescence and stabilize by adulthood.[3435] While approximately 80% of adolescents and adults with TSC have AMLs, only approximately 20% of patients with AML have TS.[36]
AMLs are the most common tumor occurring in the kidney and are typically benign. They contain variable amounts of vasculature, immature smooth muscle cells, epithelioid cells, and fat cells, all of which are deficient in either tuberin or hamartin. This deficiency may disrupt the integrated control of cell growth, leading to the AMLs.[37] Renal AMLs exhibit immunoreactivity for both melanocytic markers and smooth muscle markers.[38]
The typical appearance of an AML on CT is a noncalcified cortical mass containing fat attenuation of less than –20 HU[36] [Figure 6]. In addition to macroscopic disease, renal tissue that is radiologically normal may contain microscopic AMLs.[38] It is unclear if these microscopic lesions may grow with age.
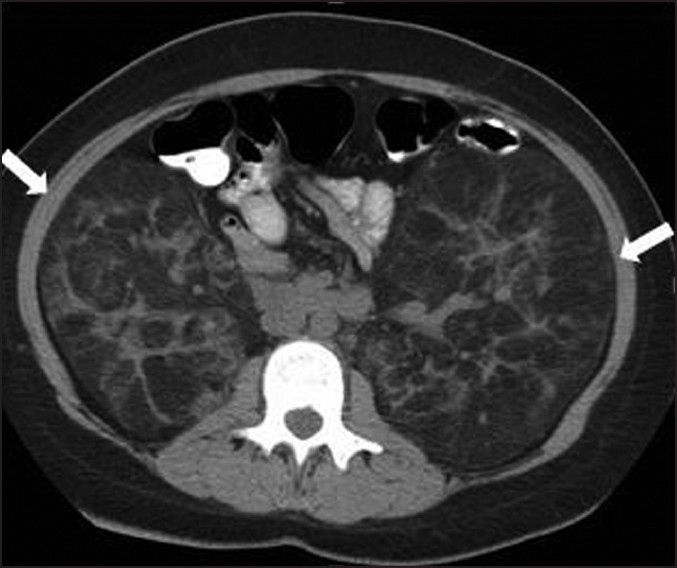
- A 45-year-old woman with tuberous sclerosis. Axial noncontrast CT image through the abdomen demonstrates enlarged kidneys with fat attenuation lesions in both kidneys consistent with angiomyolipomas
Lipid-poor lesions in patients with TSC account for 4.5–33% of AMLs.[3940] Lipid-poor lesions are defined as having less than 25% fat per high-power field,[41] on average contain 4.1% fat.[42] Lipid-poor TSC associated AML may consist of predominantly spindle cells, epithelioid cells, or vascular elements.[38] Lipid-poor AMLs are clinically relevant due to their differing clinical course and difficult distinction from renal cell carcinoma. Radiographic features that may help differentiate AML from renal cell cancer (RCC) are homogenous enhancement on CT, lack of calcifications and homogenous isoechogenicity on ultrasound.[3943] At present, there is no standard CT Hounsfield value to separate lipid poor-AML from RCC.[44–46] Use of AMLs’ growth rate has been presented as a possible method to distinguish more ominous lesions such as RCC from lipid-poor AML.[47] Utilization of MR imaging artifacts such as in-phase/out-of-phase techniques and chemical shift artifact can assist in identifying fat in lipid-poor lesions.[4849] If concern remains, PET scans can be helpful because AMLs are generally not PET avid.[5051] If there is concern for RCC and tissue diagnosis is essential, biopsy may be considered. Epithelioid AML can exhibit an aggressive phenotype, with metastasis and recurrence,[52] local invasion, including extension into renal vein or inferior vena cava,[53] and lymph node involvement.[5455] Distinction of these aggressive AMLs from renal cancer is difficult by imaging and these lesions are generally managed surgically.
The blood vessels within AMLs are abnormal with no internal elastic lamina. The smooth muscle is replaced by fibrous tissue and these vessels are rigid, tortuous, and prone to aneurysm formation and rupture,[56] making them clinically important. The hemorrhage risk of renal AML in TSC patients is between 25% and 50%.[5758]
Approximately 60% of patients with AML will be symptomatic at diagnosis, most commonly presenting with a combination of acute bleeding, flank pain, hematuria, or tender mass.[59] The risk of hemorrhage is associated with the size of the AML (greater than 4 cm) and size of the aneurysm (greater than 5 mm), the latter having higher sensitivity and specificity.[60] Control of active bleeding and prevention of re-bleeding are achieved by embolization of the aneurysm,[6162] that preserves most of the renal parenchyma[63] [Figure 7]. On angiogram in the acute setting, localizing the symptomatic aneurysm can be difficult, as active extravasation is rare and there is typically extensive abnormal vasculature. The “light bulb sign”, identified in 70% of patients treated following an acute bleed, may help in localization of the bleeding aneurysm. The light bulb sign is caused by a perianeurysmal hematoma displacing the surrounding tissue and vessels, causing a “filling defect” seen on all phases of the angiogram.[64] Corticosteroid therapy decreases subsequent postembolization syndrome.[61]
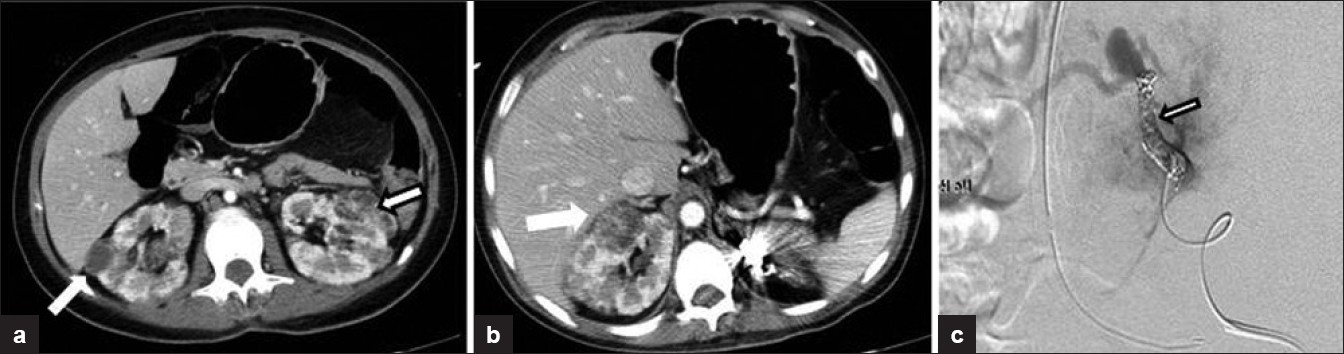
- (a-c) A 39-year-old woman with tuberous sclerosis. Axial contrast-enhanced CT images though the abdomen show multiple bilateral fat density lesions consistent with renal angiomyolipomas. Angiogram image demonstrates coiling of an aneurysm within an angiomyolipoma in the left kidney. The contrast-enhanced CT image on the right demonstrate volume loss in the left kidney following embolization
Surgical therapy is employed for a specifically targeted lesion, such as an exophytic nonbleeding AMLs, with minimal risk to the remainder of the kidney. Less invasive surgical methods such as laparoscopy, partial nephrectomy or ablative therapy with cryotherapy or radiofrequency treatment have reduced the overall impact of surgery.
Renal cystic disease
Renal cysts, usually multiple and bilateral, are the second most common kidney lesion, occurring in approximately 18–53% of patients with TSC.[86566] Although renal cysts may occasionally be present in the fetus and neonate, typically children with TSC are born with normal kidneys and develop cystic disease and AML as they age.[3234] Two percent of patients with TSC have severe early onset of polycystic phenotype associated with deletions involving adjacent TSC2 and PKD1 genes on chromosome 16p13.3.[8]
Renal cystic disease may be macrocystic, or microcystic and undetectable by imaging studies. Inactivation of the tuberin–hamartin complex is thought to result in ciliary dysfunction and defects in cell polarity leading to cyst formation.[67] In patients with TSC, cysts may arise from all segments of the nephron, including the renal tubules and the glomeruli.[6869] Animal models demonstrate acute acceleration of renal cystic disease following acute renal injury, which may also be the case in humans[70] [Figure 8a].
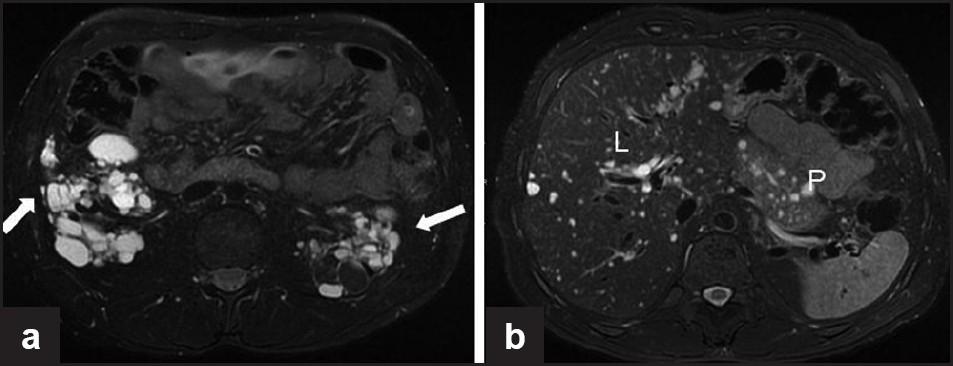
- A 33-year-old woman with tuberous sclerosis. (a) Axial T2-weighted, fat-suppressed MR image through the kidneys shows bilateral hyperintense T2 renal cysts. Also note the hypointense lesions in the left kidney in the fat-suppressed sequence, consistent with angiomyolipomas. (b) Axial T2-weighted, fat-suppressed MR image through the upper abdomen shows numerous hyperintense T2 cysts in the liver (L) and pancreas (P)
Renal cell cancer
The overall incidence of RCC in TSC patients is similar to that in the general population, with a lifetime risk of 2–3%.[171] RCC manifests at an earlier age in patients with TS (mean age 28 years) than in the general population (mean age 53 years) and shows greater histological variety with clear cell, papillary and chromophobe subtypes.[124] Growth of RCC in TSC patients is slower than sporadic RCC.[72]
CARDIAC LESIONS
In patients with TSC, cardiac rhabdomyoma [Figure 9] is the most commonly identified in utero, infancy, and early childhood. Childhood tumor regression is the rule, with most of them regressing before birth and 70% by the age of 4 years.[73] There may be a second peak in the incidence of these tumors in pubertal females with TSC.[7475] Hormonal influence of progesterone has been suggested as a cause for the bimodal distribution.
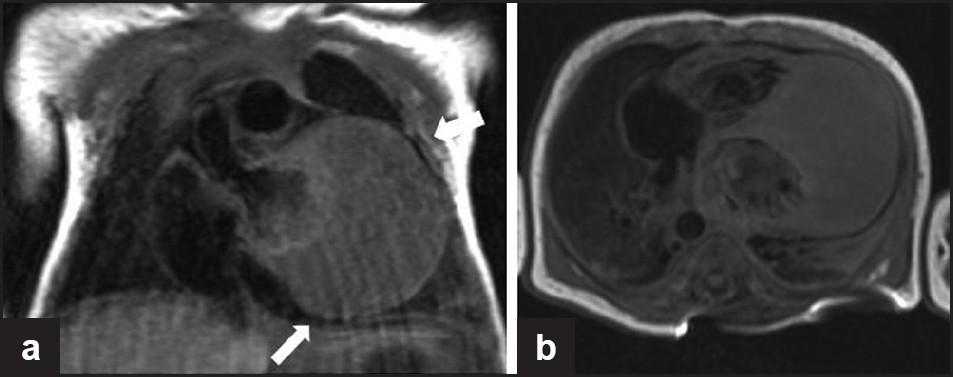
- A 2-day-old infant with arrhythmia. Cardiac gated sagittal and axial T1-weighted images show a large homogeneous isointense lesion involving the cardiac apex and the apical portions of the septum and lateral wall consistent with cardiac rhabdomyoma. This child was later diagnosed with tuberous sclerosis
These tumors are usually asymptomatic, but in a minority of cases may present with cardiac failure or arrhythmia when surgical resection may be considered. Echocardiography is commonly used in screening and follow-up of cardiac rhabdomyoma. MR imaging may provide additional information in complex cases.
PULMONARY LESIONS
The three main pulmonary lesions found in TSC are LAM, multifocal micronodular pneumocyte hyperplasia (MMPH), and clear cell “sugar” tumor (CCST) of the lung.
Lymphangioleiomyomatosis
LAM is the most common pulmonary lesion occurring essentially in only women, with the average age of onset being 33 years.[76] Pulmonary LAM is characterized by diffuse interstitial proliferation of bundles of smooth muscle cells and cystic change in pulmonary parenchyma. Dyspnea and recurrent spontaneous pneumothorax are the most common presentations, with slow and steady progression to respiratory failure.
On CT, the characteristic feature of LAM is thin-walled cysts of variable size and contour [Figure 10], which may communicate with the airways, indicated by decrease in size on expiratory imaging. In a patient with TSC, biopsy may be obviated by the presence of these characteristic findings. Occasionally, reticular opacities are identified that may represent interstitial edema due to lymphatic obstruction. Chylothorax and recurrent pneumothorax are complications which may be identified on imaging.
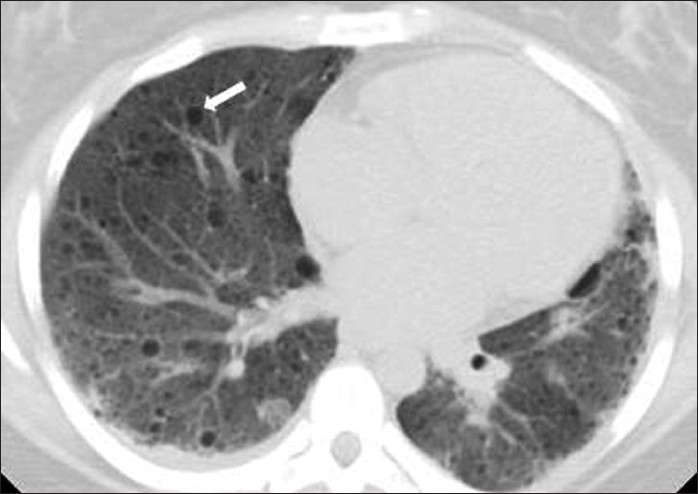
- A 24-year-old woman with tuberous sclerosis. Axial noncontrast CT image through the chest demonstrates multiple small, thin-walled, round cysts (arrow) consistent with lymphangioleiomyomatosis
In the past decade, there have been several clinical trials on hormonal therapy with progesterone and estrogen in patients with LAM, identifying downregulation of progesterone receptors.[7778] Progesterone is the most commonly used drug with a small case series and meta-analysis suggesting it to be helpful in one-third to one-half of the cases.[7980] In nonresponders, surgical intervention, such as thoracic drainage, pleurectomy or pleurodesis, may be necessary for chylothorax and recurrent pneumothorax. A combined heart and lung transplantation may be indicated for refractory LAM. Prior pleurodesis may complicate future lung transplantation.
Multifocal micronodular pneumocyte hyperplasia
MMPH is a rare disorder associated with TSC. MMPH presents as multicentric, well-demarcated hamartomatous nodules with proliferation of type II pneumocytes along the alveolar septa. MMPH also occurs predominantly in female patients and may occur with or without LAM.
On thin-section CT, MMPH is identified as multiple tiny, diffusely scattered nodules in a random distribution [Figure 11]. The appearance is nonspecific and difficult to distinguish from causes of multiple small nodules; however, in a patient with TSC, MMPH is considered with this pattern.[81] Isolated MMPH may present as dyspnea, cough, and mild to moderate hypoxemia,[82] but in view of its hamartomatous nature, it is not typically progressive or fatal.
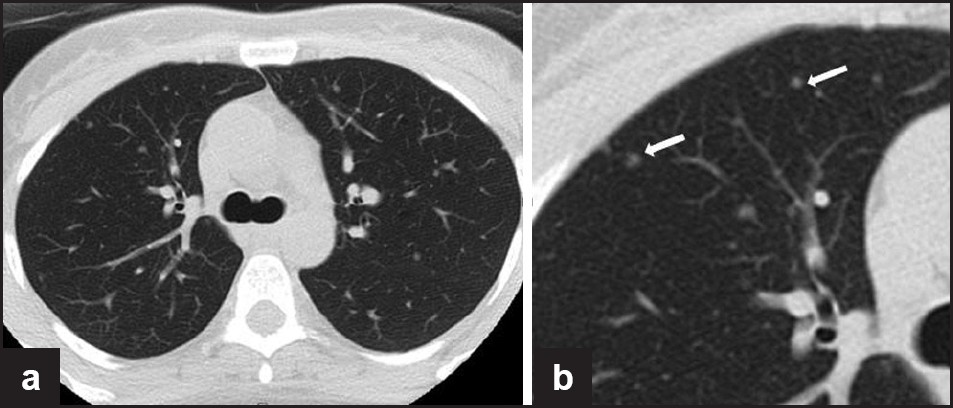
- (a, b) A 38-year-old woman with tuberous sclerosis. Axial noncontrast CT image through the chest and an enlarged view (right) demonstrating small randomly distributed soft tissue nodules, consistent with multifocal micronodular pneumocyte hyperplasia
Clear cell “sugar” tumors of the lung
CCSTs are rare benign lung lesions in TSC. These are typically unencapsulated and composed of round, oval, or focally spindled cells with distinct cellular borders, abundant clear to eosinophilic granular cytoplasm, prominent thin-walled blood channels, and focal hyaline stroma. Mitotic figures and necrosis are not usually seen.[83]
On imaging, CCSTs of the lung present as a rounded, smoothly marginated, peripheral parenchymal nodule or mass without evidence of cavitation or calcification. These lesions appear to avidly enhance on contrast-enhanced CT scans, likely related to rich vascular stroma.[8485] While CCSTs are usually benign, certain aggressive cases have been documented, occurring almost exclusively in the lung.[8687]
OTHER ABDOMINAL MANIFESTATIONS
Hepatic AMLs occur in about 13% of patients with TSC and are more common in females. Multiple hepatic AMLs may be present in patients with bilateral diffuse renal AMLs. These lesions are similar histologically to the renal AMLs.[88] On CT evaluation, hepatic AMLs are round, well-circumscribed lesions containing areas of low attenuation consistent with fat. On ultrasound, they typically present as echogenic foci. No definite follow-up has been recommended for these lesions as they are benign. Occasionally, hepatic AMLs have been reported to rupture if larger than 4 cm.[88]
Splenic hamartomas are rarely identified in patients with TSC.[89] Multiple pancreatic lesions are described, including AML.[90] Biopsy is usually recommended for pancreatic lesions.
Hamartomatous colorectal polyps are a common finding in TSC. These are usually small, less than 5 mm in diameter, and sessile or occasionally filiform in appearance.[9192]
LAM, although most common in the lungs, may also occur in other areas such as the retroperitoneum resulting in cystic lesions in the retroperitoneum and chylous ascites. Multicystic retroperitoneal lesions in a patient with TSC are considered to represent LAM. Retroperitoneal LAM can present with abdominal bloating and may cause lymphatic obstruction with lower extremity edema.
PEComas are a family of mesenchymal tumors consisting of perivascular epithelioid cells. The cell type from which these tumors originate remains unknown and has no normal counterpart. Genetically, PEComas are linked to the tuberous sclerosis genes TSC1 and TSC2. PEComas bear significant histologic and immunohistochemical similarity to AML, LAM and CCST of the lung, and CCSTs in other organs, which some authors classify under the broad category of PEComas.[93] These tumors are of perivascular epithelioid origin with a clear/granular cytoplasm and central round nucleus without prominent nucleoli. PEComas typically stain for melanocytic markers and myogenic markers and have a variable radiologic appearance. These tumors and have been reported in the falciform ligament/ligamentum teres, uterus, abdominal cavity and mesentery, soft tissue, bone, and skin [Figure 12].
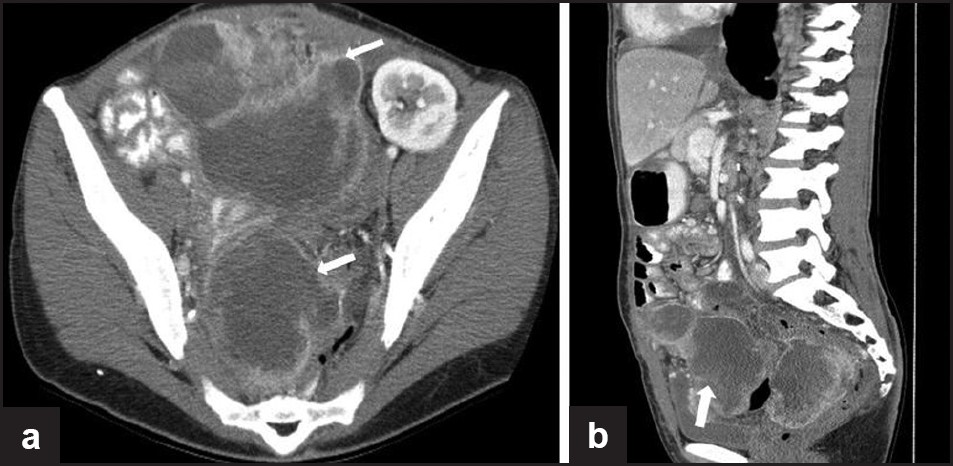
- A 35-year-old female with tuberous sclerosis and lymphangioleiomyomatosis. Status post-renal transplant. (a) Axial and (b) sagittal contrast-enhanced CT images show large, irregular, low-attenuation enhancing, peripherally enhancing masses in the pelvis in a patient with tuberous sclerosis, biopsy proven as PEComa (perivascular epitheliod cell tumor). Also note the transplant kidney in the left pelvis
SKELETAL LESIONS
Multiple bony changes have been described in TSC of which sclerotic lesions are the most common[24] [Figure 13]. Hyperostosis of inner table of skull, periosteal new bone formation, scoliosis, and bone cysts[5] have also been described.
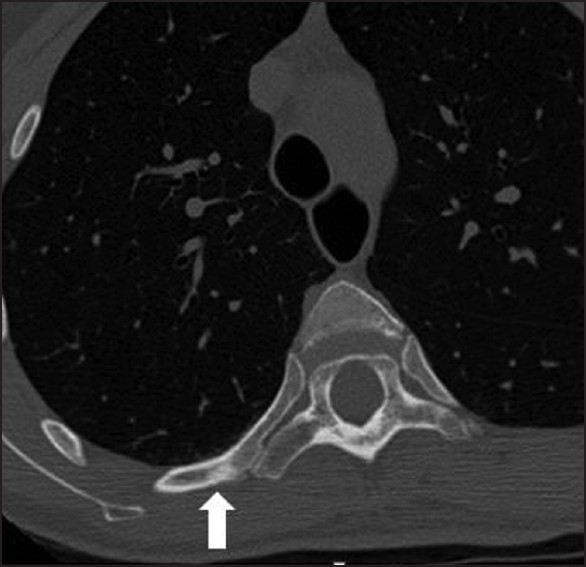
- A 30-year-old with tuberous sclerosis. Axial noncontrast chest CT demonstrates multiple small sclerotic lesions in the ribs and vertebrae
CONCLUSION
TSC is a multisystem disorder characterized by a wide spectrum of clinical and imaging features. Due to better testing and imaging methods, estimates of TSC frequency have risen dramatically in the recent years as individuals with less severe manifestations of the disorder are identified. Recognition of specific radiologic features may aid in early diagnosis and management and improve outcomes in TSC patients. Identification of patients at risk for severe manifestations of TSC and understanding the clinical impact of different radiological findings in TSC patients is crucial to improve management and outcomes.
Source of Support: Nil
Conflict of Interest: None declared.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2011/1/1/39/83230
REFERENCES
- Tuberous sclerosis complex: Neonatal deaths in three of four children of consanguineous, non-expressing parents. J Med Genet. 1997;34:256-60.
- [Google Scholar]
- Tuberous sclerosis complex consensus conference: Revised clinical diagnostic criteria. J Child Neurol. 1998;13:624-8.
- [Google Scholar]
- Linkage of an important gene locus for tuberous sclerosis to a chromosome 16 marker for polycystic kidney disease. Nat Genet. 1992;2:37-41.
- [Google Scholar]
- Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277:805-8.
- [Google Scholar]
- Renal cystic disease in tuberous sclerosis: Role of the polycystic kidney disease 1 gene. Am J Hum Genet. 1997;61:843-51.
- [Google Scholar]
- Comprehensive mutation analysis of TSC1 and TSC2-and phenotypic correlations in 150 families with tuberous sclerosis. Am J Hum Genet. 1999;64:1305-15.
- [Google Scholar]
- Mutational analysis of the TSC1 and TSC2 genes in a diagnostic setting: Genotype--phenotype correlations and comparison of diagnostic DNA techniques in Tuberous Sclerosis Complex. Eur J Hum Genet. 2005;13:731-41.
- [Google Scholar]
- Tuberous sclerosis: A GAP at the crossroads of multiple signaling pathways. Hum Mol Genet. 2005;14:R251-8.
- [Google Scholar]
- Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am J Hum Genet. 1996;59:400-6.
- [Google Scholar]
- Loss of tuberin in both subependymal giant cell astrocytomas and angiomyolipomas supports a two-hit model for the pathogenesis of tuberous sclerosis tumors. Am J Pathol. 1997;151:1639-47.
- [Google Scholar]
- Russel and Rubensten's Pathology of Tumors of the Nervous System. (7th ed). London: Hodder Arnold Publication; 2006.
- [Google Scholar]
- Subependymal nodules, giant cell astrocytomas and the tuberous sclerosis complex: A population-based study. Arch Dis Child. 2008;93:751-4.
- [Google Scholar]
- Cystlike white matter lesions in tuberous sclerosis. AJNR Am J Neuroradiol. 1997;18:1367-73.
- [Google Scholar]
- MR imaging of tuberous sclerosis: Pathogenesis of this phakomatosis, use of gadopentetate dimeglumine, and literature review. Radiology. 1992;183:227-38.
- [Google Scholar]
- Tuberous sclerosis in the fetus: Second-trimester diagnosis of subependymal tubers with ultrafast MR imaging. AJR Am J Roentgenol. 2000;175:1067-9.
- [Google Scholar]
- Transmantle dysplasia in tuberous sclerosis: Clinical features and surgical outcome in four children. J Child Neurol. 2002;17:752-8.
- [Google Scholar]
- Pictorial review of tuberous sclerosis in various organs. Radiographics. 2008;28:e32.
- [Google Scholar]
- Brain lesions in tuberous sclerosis complex.Review. Folia Neuropathol. 2010;48:139-49.
- [Google Scholar]
- Cortical tubers, cognition, and epilepsy in tuberous sclerosis. Pediatr Neurol. 2011;44:328-32.
- [Google Scholar]
- Cortical tuber count: A biomarker indicating neurologic severity of tuberous sclerosis complex. J Child Neurol. 1997;12:85-90.
- [Google Scholar]
- MRI findings reveal three different types of tubers in patients with tuberous sclerosis complex. J Neurol. 2010;257:1373-81.
- [Google Scholar]
- Sequential CT study of subependymal giant-cell astrocytoma associated with tuberous sclerosis.Case report. J Neurosurg. 1986;65:874-7.
- [Google Scholar]
- Subependymal giant cell astrocytoma with high choline/creatine ratio on proton MR spectroscopy. Arq Neuropsiquiatr. 2006;64:877-80.
- [Google Scholar]
- Causes of death in patients with tuberous sclerosis. Mayo Clin Proc. 1991;66:792-6.
- [Google Scholar]
- Renal lesion growth in children with tuberous sclerosis complex. J Urol. 1998;160:141-5.
- [Google Scholar]
- Tuberous sclerosis complex developmental perspectives in psychiatry. New York: Oxford University press; 1999.
- [Google Scholar]
- The relation of infantile spasms, tubers, and intelligence in tuberous sclerosis complex. Arch Dis Child. 2004;89:530-3.
- [Google Scholar]
- Best cases from the AFIP: Angiomyolipomas in tuberous sclerosis. Radiographics. 2003;23:241-6.
- [Google Scholar]
- Clinical and molecular insights into tuberous sclerosis complex renal disease. Pediatr Nephrol. 2011;26:839-52.
- [Google Scholar]
- Angiomyolipoma with minimal fat: Differentiation from renal cell carcinoma at biphasic helical CT. Radiology. 2004;230:677-84.
- [Google Scholar]
- CT and MR imaging of extrahepatic fatty masses of the abdomen and pelvis: Techniques, diagnosis, differential diagnosis, and pitfalls. Radiographics. 2005;25:69-85.
- [Google Scholar]
- Fat poor renal angiomyolipoma: Patient, computerized tomography and histological findings. J Urol. 2006;176:905-9.
- [Google Scholar]
- Imaging characteristics of minimal fat renal angiomyolipoma with histologic correlations. Urology. 2005;66:1155-9.
- [Google Scholar]
- Angiomyolipoma: Imaging findings in lesions with minimal fat. Radiology. 1997;205:497-502.
- [Google Scholar]
- Diagnosis of angiomyolipoma using computed tomography-region of interest < or =-10 HU or 4 adjacent pixels < or =-10 HU are recommended as the diagnostic thresholds. Clin Radiol. 2006;61:410-6.
- [Google Scholar]
- CT histogram analysis: Differentiation of angiomyolipoma without visible fat from renal cell carcinoma at CT imaging. Radiology. 2008;246:472-9.
- [Google Scholar]
- Clinical correlates of renal angiomyolipoma subtypes in 209 patients: Classic, fat poor, tuberous sclerosis associated and epithelioid. J Urol. 2008;180:836-43.
- [Google Scholar]
- Tuberose sclerosis complex: Analysis of growth rates aids differentiation of renal cell carcinoma from atypical or minimal-fat-containing angiomyolipoma. Clin Radiol. 2005;60:665-73. discussion 663-4
- [Google Scholar]
- Benign adrenocortical masses: Diagnosis with chemical shift MR imaging. Radiology. 1992;185:345-51.
- [Google Scholar]
- The use of opposed-phase chemical shift MRI in the diagnosis of renal angiomyolipomas. AJR Am J Roentgenol. 2005;184:1868-72.
- [Google Scholar]
- Utility of [18F]2-fluoro-2-deoxyglucose-PET in sporadic and tuberous sclerosis-associated lymphangioleiomyomatosis. Chest. 2009;136:926-33.
- [Google Scholar]
- The tuberous sclerosis complex regulates trafficking of glucose transporters and glucose uptake. Am J Pathol. 2008;172:1748-56.
- [Google Scholar]
- Epithelioid angiomyolipoma of the kidney: A report of five cases with a prominent and diagnostically confusing epithelioid smooth muscle component. Am J Surg Pathol. 1997;21:1123-30.
- [Google Scholar]
- Case report: Invasive renal angiomyolipoma--sonographic and CT features. Clin Radiol. 1993;48:283-5.
- [Google Scholar]
- Trends of presentation and clinical outcome of treated renal angiomyolipoma. Yonsei Med J. 2010;51:728-34.
- [Google Scholar]
- The tuberous sclerosis complex and its highly variable manifestations. J Urol. 2003;169:1635-42.
- [Google Scholar]
- Symptomatic renal angiomyolipoma: Report of 8 cases, 2 with spontaneous rupture. J Urol. 1978;119:684-8.
- [Google Scholar]
- Contemporary diagnosis and management of renal angiomyolipoma. J Urol. 2002;168:1315-25.
- [Google Scholar]
- Renal angiomyolipoma: Relationships between tumor size, aneurysm formation, and rupture. Radiology. 2002;225:78-82.
- [Google Scholar]
- Reduction of postembolization syndrome after ablation of renal angiomyolipoma. Am J Kidney Dis. 2002;39:966-71.
- [Google Scholar]
- Embolization of renal angiomyolipomata in patients with tuberous sclerosis complex. Am J Kidney Dis. 2006;47:95-102.
- [Google Scholar]
- Long-term outcome of transcatheter embolization of renal angiomyolipomas due to tuberous sclerosis complex. J Urol. 2005;174:1764-6.
- [Google Scholar]
- Embolization of renal angiomyolipoma: Immediate complications and long-term outcomes. Clin Radiol. 2008;63:864-70.
- [Google Scholar]
- Renal manifestations of tuberous sclerosis complex: Incidence, prognosis, and predictive factors. Kidney Int. 2006;70:1777-82.
- [Google Scholar]
- The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum Mol Genet. 2009;18:151-63.
- [Google Scholar]
- Diffuse bilateral glomerulocystic disease of the kidneys and multiple cardiac rhabdomyomas in a newborn.Relationship with tuberous sclerosis and review of the literature. Pathol Res Pract. 1992;188:367-73. discussion 373-4
- [Google Scholar]
- Acute kidney injury and aberrant planar cell polarity induce cyst formation in mice lacking renal cilia. Hum Mol Genet. 2008;17:1578-90.
- [Google Scholar]
- Magnetic resonance imaging of renal involvement in genetically studied patients with tuberous sclerosis complex. Eur J Radiol. 2009;72:335-41.
- [Google Scholar]
- Evolution of cardiac rhabdomyoma in tuberous sclerosis complex. Clin Pediatr (Phila). 1996;35:615-9.
- [Google Scholar]
- Cardiac tumours in tuberous sclerosis: Their incidence and course. Eur J Pediatr. 1994;153:155-7.
- [Google Scholar]
- Fetal rhabdomyoma: Two instances of recurrence. Pediatr Pathol Lab Med. 1996;16:673-80.
- [Google Scholar]
- Downregulation of estrogen and progesterone receptors in the abnormal smooth muscle cells in pulmonary lymphangioleiomyomatosis following therapy.An immunohistochemical study. Am J Respir Crit Care Med. 2000;161:1002-9.
- [Google Scholar]
- Frequent estrogen and progesterone receptor immunoreactivity in renal angiomyolipomas from women with pulmonary lymphangioleiomyomatosis. Chest. 2000;117:25-30.
- [Google Scholar]
- Pulmonary lymphangioleiomyomatosis: A report of 46 patients including a clinicopathologic study of prognostic factors. Am J Respir Crit Care Med. 1995;151:527-33.
- [Google Scholar]
- Multifocal micronodular pneumocyte hyperplasia in tuberous sclerosis. AJR Am J Roentgenol. 2005;184:S37-9.
- [Google Scholar]
- Multifocal micronodular pneumocyte hyperplasia and lymphangioleiomyomatosis in tuberous sclerosis with a TSC2 gene. Mod Pathol. 2001;14:609-14.
- [Google Scholar]
- Clear cell “sugar” tumor of the lung: Association with lymphangioleiomyomatosis and multifocal micronodular pneumocyte hyperplasia in a patient with tuberous sclerosis. Am J Surg Pathol. 1997;21:1242-7.
- [Google Scholar]
- A rare cause of hemoptysis: benign sugar (clear) cell tumor of the lung. Eur J Cardiothorac Surg. 2004;25:652-4.
- [Google Scholar]
- Malignant clear cell sugar tumor of the lung: Patient case report. J Clin Oncol. 2010;28:e626-8.
- [Google Scholar]
- ‘Benign’ clear-cell tumor (sugar tumor) of the lung with hepatic metastases ten years after resection of pulmonary primary tumor. Arch Pathol Lab Med. 1988;112:1177-8.
- [Google Scholar]
- Frequency and imaging appearance of hepatic angiomyolipomas in pediatric and adult patients with tuberous sclerosis. AJR Am J Roentgenol. 2004;182:1027-30.
- [Google Scholar]
- Extrathoracic angiomyolipomas in lymphangioleiomyomatosis. Eur Respir J. 1996;9:402-5.
- [Google Scholar]
- Hamartomatous rectal polyps are common in tuberous sclerosis. Ann N Y Acad Sci. 1991;615:71-80.
- [Google Scholar]




