
Translate this page into:
Waardenburg Syndrome and Left Persistent Superior Vena Cava
Address for correspondence: Dr. Alexander Christie, Department of Radiology, University of Kentucky, Kentucky, USA. E-mail: adch223c@gmail.com
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Waardenburg syndrome (WS) is a rare genetic disorder secondary to neural crest cell developmental abnormalities. It is predominantly described as an auditory–pigmentary syndrome with diverse patient presentation, typically involving congenital sensorineural hearing loss and pigmentation abnormalities of the skin, hair, and iris. Other developmental abnormalities that may be associated with this syndrome are Hirschsprung's disease and a myriad of cardiovascular congenital defects. We present a case of a young girl with WS who found to have a persistent left superior vena cava (PLSVC) draining into the coronary sinus. The prevalence of PLSVC is increased in patients with chromosomal and genetic abnormalities. However, we are the first to report its presence in association with WS while discussing the challenges that may arise during central venous catheter placement in patients with PLSVC.
Keywords
Central venous anomalies
duplicated superior vena cava
Hirschsprung's disease
left superior vena cava
Waardenburg syndrome


INTRODUCTION
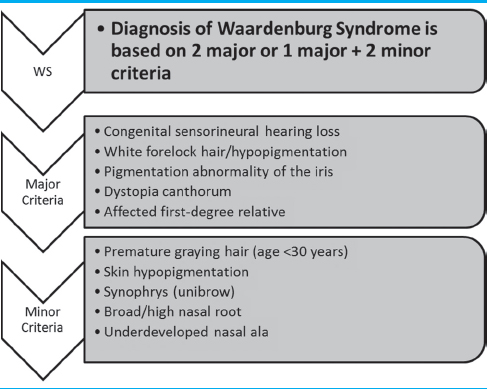
Waardenburg syndrome (WS) is a rare but clinically important inherited genetic disorder, as it is estimated to be responsible for 2%–5% of congenital deafness.[1] It is predominantly described as an auditory–pigmentary syndrome, with sensorineural hearing loss and pigmentary changes being the most prominent characteristics.[1] It is also classified among neurocutaneous syndromes such as neurofibromatosis, Sturge–Weber syndrome, and tuberous sclerosis. There are four main subtypes of WS with varying modes of inheritance and expression, with autosomal dominant being the most common.[1] The diagnosis is made clinically based on the presence of major and minor criteria involving multiple signs and symptoms [Table 1]. Some common criteria include congenital sensorineural hearing loss, iris pigmentary abnormalities (most commonly heterochromia iridis), abnormalities of hair pigmentation, and dystopia canthorum. Dystopia canthorum is the classic facial appearance that mimics the visual appearance of hypertelorism but actually consists of the lateral displacement of the inner canthi of the eyes. The clinical diagnosis can be confirmed by genetic testing. The pathogenesis of WS involves multiple possible mutations in genes responsible for the development of neural crest cells. The resulting defects in melanocytes are responsible for the hypopigmentary changes of the skin and the classic forelock of white hair seen in WS Type-I.
Considering the abnormalities in neural crest migration in WS, one of the most clinically significant associations is intestinal aganglionosis or Hirschsprung's disease. It is specifically present in WS Type-IV. Multiple other developmental defects have been described in association with WS, namely limb abnormalities, congenital renal anomalies, Dandy–Walker malformation, and several cardiovascular malformations.
In the cardiovascular system, there have been descriptions of inferior vena cava interruption and congenital heart disease, including severe cyanotic cardiomyopathy and atrial septal defect.[2] This diverse set of associated cardiovascular anomalies can be traced to the embryology involving neural crest cells. Neural crest cell migration is responsible for the development of many structures deriving from ectodermal and mesodermal origins. Similar to melanocytes, connective tissue, and most of the ocular structures, blood vessels trace their origin to the mesoderm.
The early diagnosis of hearing impairment is key to improve the outcome of patients with WS; thus, imaging of the temporal bone and inner ear plays a major role in the workup of WS patients prior to cochlear implantation. WS can be associated with a myriad of cochlear, semicircular canal, and temporal bone malformations described on computed tomography and magnetic resonance imaging.[3] Furthermore, since Hirschsprung's disease has well-described radiological findings and is known to have a strong association with WS, the pediatric radiologist should be cognizant of this potential association. Despite these various malformations and the aforementioned cardiovascular anomalies, the presence of a persistent left superior vena cava (PLSVC) in a patient with WS has yet to be described.
CASE REPORT
We report the case of a 5-year-old girl with a history of WS Type-II associated with left Bochdalek diaphragmatic hernia, dysmorphic facies, and hypoxia. She also had a history of cecal volvulus with resulting bowel infarction and resection. She underwent repeated laparotomies for bowel ischemia and abdominal compartment syndrome, resulting in short gut syndrome and requiring total parental nutrition. For this indication, the interventional radiology service was consulted for peripherally inserted central catheter (PICC) placement. The patient had multiple central lines placed in the past and was considered to have difficult venous access.
During the PICC placement procedure, access was initially obtained in the right basilic vein, but there was difficulty in advancing the wire distally. Right upper-extremity contrast venography demonstrated chronic total occlusion of the right subclavian vein, likely the result of prior indwelling central venous catheters [Figure 1]. Venous access was attempted on the left brachial vein. The wire advanced along the left paraspinal line and would not cross the mid sternal line to the right atrium. Left upper-extremity contrast venography was performed [Figure 2], showing a PLSVC draining into the right atrium via the coronary sinus. The Single-Lumen 3 French PICC was then advanced over the wire and imaged in the coronary sinus before retraction and successful positioning in the left superior vena cava [Figure 3]. Three months later, there was concern for an infectious process and CT scan of the chest was performed. The PLSVC is redemonstrated [Figures 4 and 5].
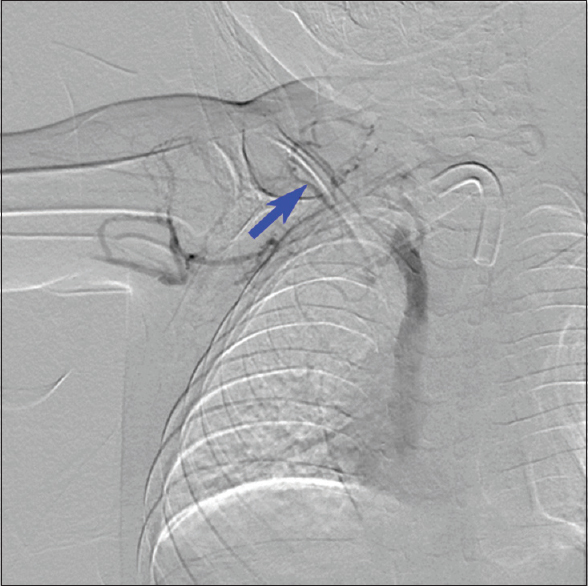
- A 5-year-old female with Waardenburg syndrome presented for peripherally inserted central catheter placement for total parental nutrition. Digital subtraction venography of the right upper extremity showing chronic total occlusion of the right axillary and subclavian veins (blue arrow).
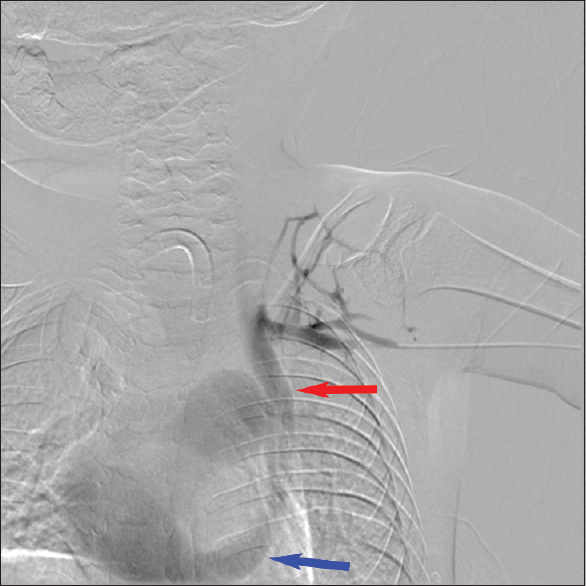
- Digital subtraction venography of the left upper extremity showing patient's cephalic arch and subclavian vein draining into a left-sided superior vena cava (red arrow). Left-sided superior vena cava drains to the right atrium via the coronary sinus (blue arrow).
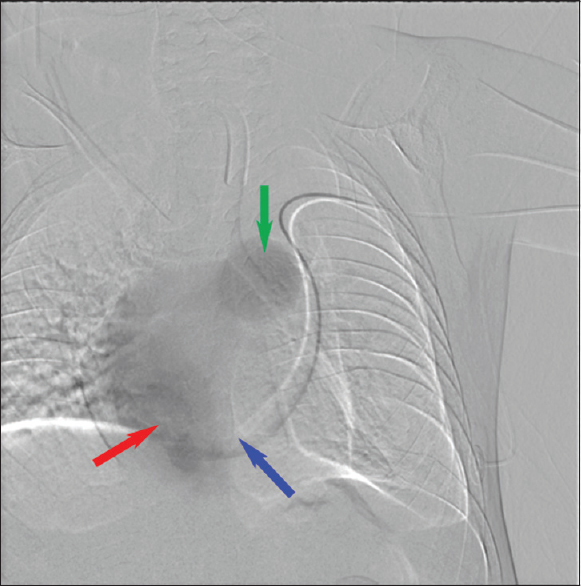
- Peripherally inserted central catheter line catheter tip (blue arrow) venogram within the coronary sinus (red arrow) showing the right atrium and the main pulmonary artery truck (green arrow).
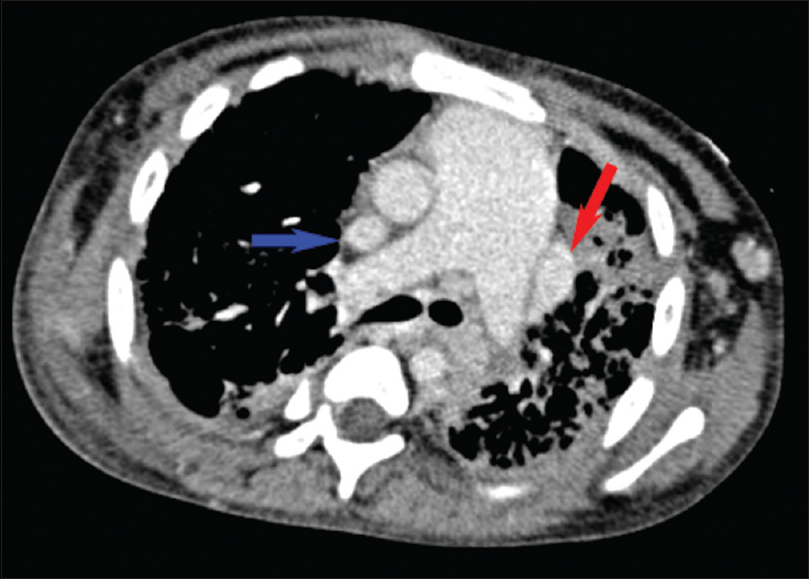
- Axial contrast-enhanced chest computed tomography showing duplicated superior vena cava (blue and red arrows) and an enlarged pulmonary artery trunk.
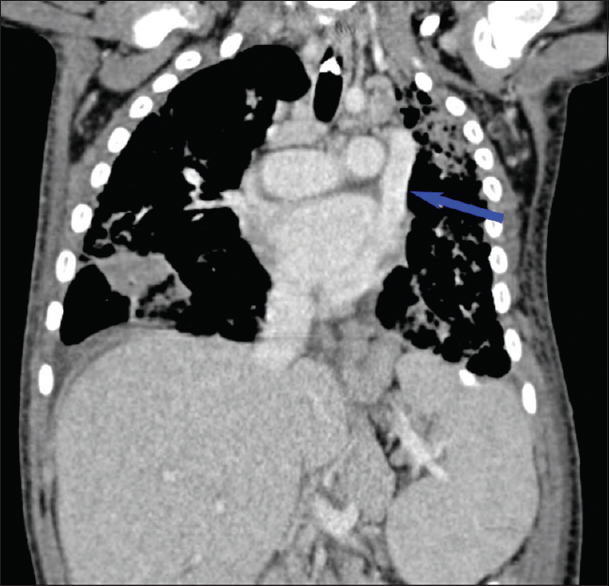
- Coronal contrast-enhanced chest computed tomography showing a left-sided superior vena cava (blue arrow).
DISCUSSION
PLSVC is the most common congenital venous anomaly in the chest and is estimated to be present in 0.3%–0.5% of the general population, although its prevalence could be higher due to its largely asymptomatic nature. In 92% of cases, its venous drainage course involves drainage into the right atrium via the coronary sinus.[4] In the remainder of cases, the PLSVC drains directly into the left atrium, resulting in a right-to-left shunt. Even in these cases, however, patients can be asymptomatic as the shunt is usually not large enough to cause cyanosis. However, PLSVC is associated with congenital heart defects in up to 40% of cases.[4]
While the presence of PLSVC is rare in the general population, it has been described as more prevalent in patients with chromosomal and genetic anomalies.[5] The embryological origin of PLSVC stems from failure of regression of the left anterior cardinal vein.[5] During the development of the thoracic vasculature, an anastomosis forms that joins the right and left brachiocephalic veins. If this does not occur, the left anterior cardinal vein retains its connection with the left horn of the sinus venosus. The end result is a PLSVC that communicates with the coronary sinus. Given the previously described defects in neural crest cell migration in WS, there is the potential for developmental abnormalities in the vasculature arising from the mesoderm.
Our patient's diagnosis of PLSVC involved the typical course of drainage into the coronary sinus and was therefore asymptomatic from the PLSVC standpoint. In the neonatal period, she was thought to have congenital heart disease given the prominent dilated appearance of the main pulmonary artery and a suspected atrial septal defect (later identified as a patent foramen ovale). Interestingly, our patient did not have congenital heart defects accompanying her PLSVC as would have been expected, and echocardiographic evaluation demonstrated that her dilated pulmonary artery was present in the absence of pulmonary hypertension.
The most common indication for PICC placement in the pediatric population includes the need for long-term chemotherapy, antibiotics, and total parenteral nutrition. It is well accepted that the ideal catheter tip position for a PICC is between the carina and up to two vertebral bodies below it on chest X-ray (CXR) or fluoroscopy. Catheter tip location beyond that range can be associated with increased catheter-related complications. For procedures not done under fluoroscopy, the successful placement of any central line requires a follow-up CXR. In our case, a left paraspinal catheter tip position was identified under fluoroscopy, prompting immediate workup. The differential of a left paraspinal central venous catheter tip includes extravascular placement, arterial placement in the thoracic aorta, and multiple venous positions including placement in the PLSVC, the left pericardiophrenic vein, and left internal thoracic vein. Considering the various implications of a left paraspinal central venous catheter tip, prior knowledge of the patient's history and comfort with this differential could avoid further imaging or intervention. After an extensive English language literature review involving search keywords of “Waardenburg syndrome” with association of “Persistent left superior vena cava,” “Duplicate superior vena cava,” “Vascular anomalies,” “Cardiovascular anomalies,” and “Congenital heart disease” using PubMed, Google Scholar, and Cochrane database, we concluded that this is the first reported case of WS in association with PLSVC. Its incidental discovery during a peripherally inserted central venous catheter highlights the challenges that can arise during central venous catheter placement in the pediatric population. A high index of suspicion coupled with good knowledge of the left paraspinal central venous catheter tip differential diagnosis can be of paramount importance.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2018/8/1/44/245523
REFERENCES
- Waardenburg's syndrome and severe cyanotic cardiopathy. Arch Fr Pediatr. 1990;47:657-9.
- [Google Scholar]
- Inner ear anatomy in Waardenburg syndrome: Radiological assessment and comparison with normative data. Int J Pediatr Otorhinolaryngol. 2014;78:1320-6.
- [Google Scholar]
- Persistent left superior vena cava: Case report and literature review. Respir Care. 2000;45:411-6.
- [Google Scholar]
- Fetal persistent left superior vena cava in cases with and without chromosomal anomalies. Prenat Diagn. 2014;34:797-802.
- [Google Scholar]




