
Translate this page into:
Two Cases of Spinal, Extraosseous, Intradural Ewing's sarcoma/Peripheral Neuroectodermal Tumor: Radiologic, Pathologic, and Molecular Analysis
Address for correspondence: Dr. Mark T Curtis, Room 282 Main Building, Department of Pathology, Thomas Jefferson University Hospital, 132 South 10th Street, Philadelphia, PA 19107, United States. E-mail: mark.curtis@jefferson.edu
-
Received: ,
Accepted: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Extraosseous Ewing's sarcoma/peripheral neuroectodermal tumors (ES/PNETs) are rare neoplasms that account for approximately 10%-15% of soft tissue sarcomas in children and 5% of soft tissue sarcomas in adults. Primary spinal, extraosseous, intradural ES/PNETs are even less common. The diagnosis of ES/PNET is extremely challenging, because the tumor can have a nonspecific radiologic appearance, and the histologic features are shared by many other “small round cell tumors.” Thus, ES/PNET should be included in the radiologic and pathologic differential diagnosis, even in older patients and in unusual tumor sites. We report two cases of spinal, extraosseous, intradural ES/PNETs in adults who presented with back pain. Magnetic resonance imaging revealed contrast enhancing, intradural lesions in the area of the conus medullaris. The tumor in Case 1 was partially intramedullary, while the tumor in Case 2 was exclusively extramedullary. In both cases, the radiologic and intraoperative surgical impression favored ependymoma. The diagnosis of ES/PNET was established in both cases by histopathologic, immunohistochemical, and molecular analysis.
Keywords
Ewing sarcoma/peripheral neuroectodermal tumor
intradural spinal neoplasms
peripheral neuroectodermal tumors

INTRODUCTION

Ewing's sarcoma/peripheral neuroectodermal tumor (ES/PNET) is a highly malignant primitive neoplasm, occurring most commonly as a primary tumor of bone in children and young adults.[1] When ES/PNET affects older patients, the tumor most often presents as an extraosseous soft tissue mass.[2]
ES/PNET is a member of a group of heterogenous mesenchymal tumors with round cell morphology designated “small round cell sarcomas,” which includes alveolar rhabdomyosarcoma, desmoplastic small round cell tumor, mesenchymal chondrosarcoma, and poorly differentiated synovial sarcoma.[3] Other tumors that may have a similar “small round cell” appearance include lymphoma, Merkel cell carcinoma, small cell carcinoma, and Wilms’ tumor. A precise diagnosis is critical, as the management of ES/PNET differs dramatically from the other histologically similar neoplasms.
We present two cases of ES/PNET occurring in the spinal, intradural location. A small number of cases of ES/PNET have been reported in this unusual location.[4] Case 1 was a 26-year-old male with a recent history of back pain. Case 2 was a 70-year-old male with increasing back pain and a history of multiple tumors. The tumor in Case 1 was shown by radiologic and intraoperative examination to be intramedullary, an extremely rare occurrence, while the tumor in Case 2 was extramedullary. These two cases highlight the importance of considering ES/PNET in the radiologic differential for intradural spinal tumors.
RADIOLOGIC FEATURES
A 26-year-old male (Case 1) presented with new onset of low back pain without antecedent injury. Magnetic resonance imaging (MRI) of the lumbar spine showed a 4.1 × 1.6 cm, T1 hypointense, and T2 hyperintense, intradural mass lesion at T12-L1, arising from the area of the conus medullaris and extending inferiorly. The lesion was densely invested with the roots of the cauda equina and demonstrated heterogeneous enhancement [Figure 1]. There was no evidence of hemorrhage, peritumoral edema, or syringomyelia. Imaging of the entire central nervous system showed no evidence of leptomeningeal enhancement or cerebrospinal fluid (CSF) seeding. With these radiologic features and tumor location, ependymoma was considered the most likely diagnosis. Other tumors in the differential, including schwannoma and metastasis, were considered less likely.
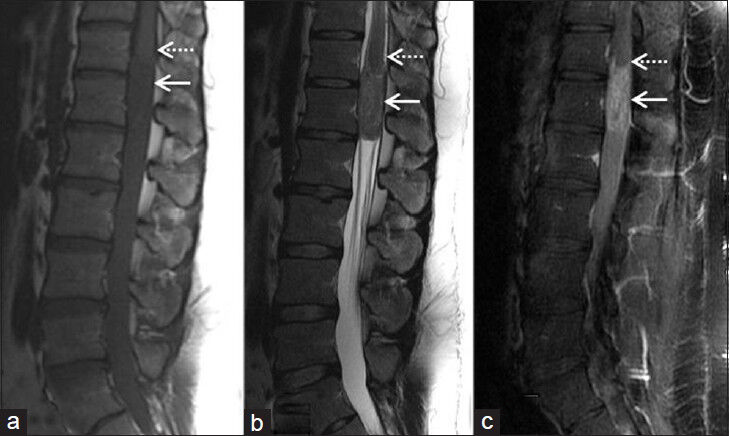
- Case 1. 26-year-old male with low back pain diagnosed with Ewing's sarcoma/peripheral neuroectodermal tumor. (a) Magnetic resonance imaging (MRI) sagittal T1 WI image shows a hypointense intradural mass lesion (solid arrow) at the level of the conus medullaris (dashed arrow), (b) MRI Sagittal T2 WI image shows the lesion (solid arrow) to be mildly hyperintense, and (c) MRI sagittal postcontrast T1 WI image shows the lesion (solid arrow) demonstrates heterogeneous enhancement after administration of contrast. The conus cannot be visualized separately from the lesion in all images (dashed arrows).
A 70-year-old male (Case 2) with a history of multiple tumors, including prostate cancer, colon cancer, gastric cancer, and right arm schwannoma, presented with worsening back pain. MRI of the lumbar spine showed a 3.0 × 1.7 cm intradural extramedullary mass at T12-L1, displacing the spinal cord to the left. The lesion was predominantly hypointense on T1, heterogeneously hyperintense on T2 with a cystic area. The lesion demonstrated heterogeneous enhancement [Figure 2]. There was no signal abnormality in the adjacent spinal cord or syringomyelia. There was no evidence of secondary lesion or leptomeningeal enhancement in the brain, thoracic and upper cervical spine. Ependymoma was the most probable diagnosis, but in view of the patient's history of malignancy, the possibility of metastasis was also considered. Although the patient had a history of schwannoma in the upper extremity, this diagnosis was considered less likely.
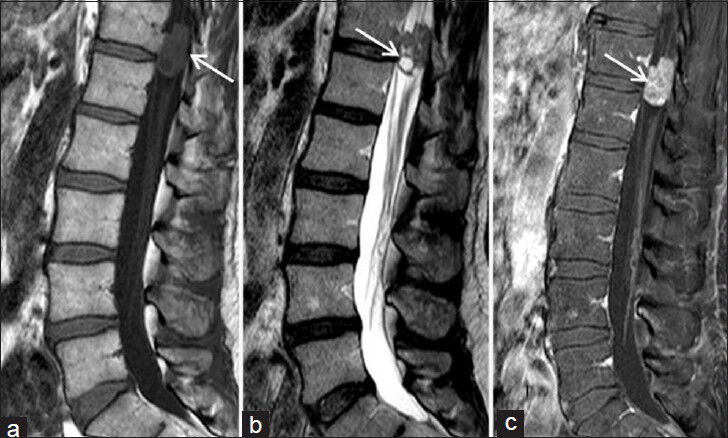
- Case 2. 70-year-old male with increasing back pain diagnosed with Ewing's sarcoma/peripheral neuroectodermal tumor. (a) Magnetic resonance imaging (MRI) sagittal T1 WI image shows a hypointense intradural extramedullary mass (arrow) at the level of T12-L1, (b) MRI Sagittal T2 WI image shows a mixed signal intensity, predominantly hyperintense signal mass with a cystic component (arrow), and (c) MRI sagittal postcontrast T1 WI image demonstrates heterogeneous enhancement of the lesion (arrow).
PATHOLOGIC FEATURES
The intraoperative impression in Case 1 was that the mass was grossly consistent with ependymoma. The tumor was noted to have both an intramedullary and an extramedullary component. The extramedullary portion of the tumor was completely resected first, and then the intramedullary portion of the tumor was gently dissected off the rootlets with eventual resection of all identified tumor. Intraoperative gross examination in Case 2 revealed a purple-red lobulated mass. Due to extensive entanglement of nerve roots in the tumor, the surgical goal was to debulk as much of the mass as possible while avoiding damage to peripheral nerve function.
Histologic examination of both tumors showed densely packed sheets and nests of small cells with scant cytoplasm and round to ovoid nuclei [Figure 3]. No microscopic features of ependymoma, such as perivascular pseudorosettes or ependymal rosettes, were identified in either case. Due to the nonspecific small blue cell appearance of these tumors, a series of immunohistochemical analyses was performed in each case. The results and interpretation of the various stains are shown in Table 1. The immunohistochemical analysis ruled out lymphoma, Merkel cell carcinoma, rhabdomyosarcoma, ependymoblastoma, neuroendocrine carcinoma, Wilms’ tumor, germ cell tumor, and prostate cancer. These findings, together with positive staining for cluster of differentiation 99 (CD99) and Friend leukemia integration 1 transcription factor (FLI-1) in Case 1 and positive staining for CD99 in Case 2, increased the likelihood that these tumors were both ES/PNETs.
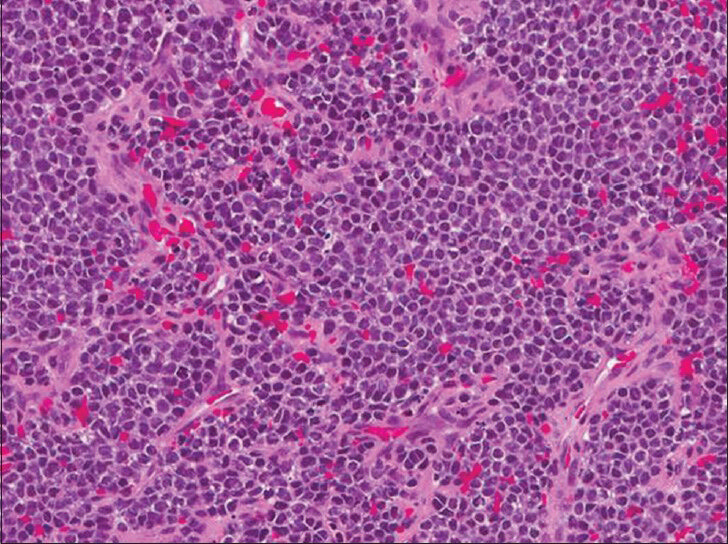
- Hematoxylin and eosin stained sections of tumors from both cases (Case 2 shown here) show the classic histologic appearance of a small blue cell neoplasm with nests of small round tumor cells with scanty cytoplasm.
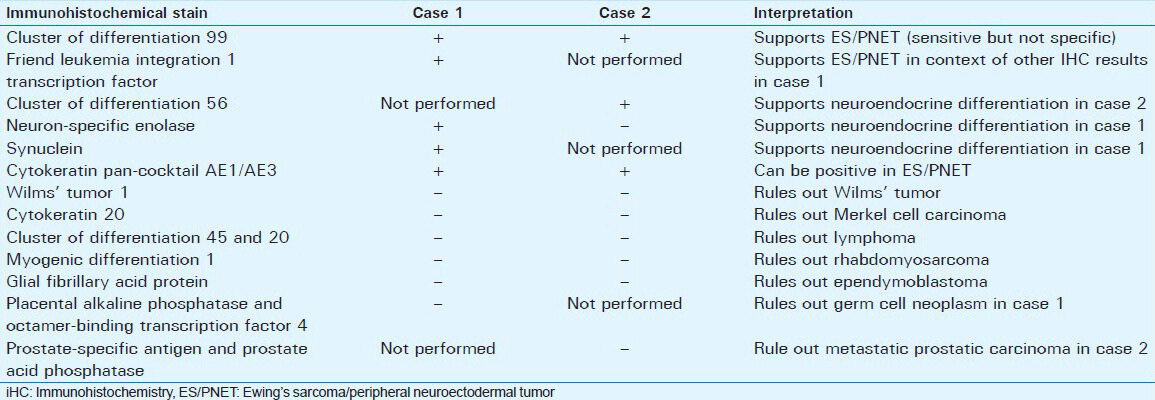
Over 90% of ES/PNETs have a chromosomal translocation involving the Ewing's sarcoma breakpoint region 1 (ESWR1).[5] Fluorescent in-situ hybridization (FISH) break-apart assay for the EWSR1 gene rearrangement was performed and revealed that both cases were positive for this rearrangement, confirming the diagnosis of ES/PNET [Figure 4]. In an attempt to identify a specific translocation, both cases were tested using reverse transcription polymerase chain reaction (RT-PCR). No specific translocation was detected by the RT-PCR assays performed on the tumor in Case 1. The tumor in Case 2 was positive for the presence of the Ewing sarcoma gene-FLI1 gene (EWS-FLI1) fusion transcript associated with ES/PNET.
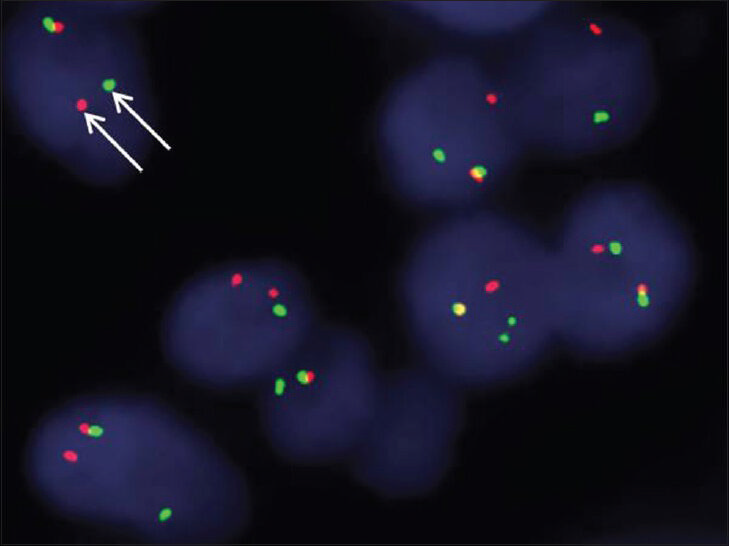
- Fluorescent in situ hybridization for Ewing's sarcoma breakpoint region 1 (ESWR1) gene rearrangement performed reveals that the tumors in both cases (Case 2 shown here) are positive for EWSR1 gene rearrangement as indicated by the separation of red and green signals (arrows).
DISCUSSION
ES/PNETs are aggressive, malignant neoplasms that share histologic features with a number of other tumors. Multimodality therapy with surgery, radiation therapy, and chemotherapy has improved survival in ES/PNET from less than 10% to up to 40%.[6] Immunohistochemical analysis of small round cell tumors is crucial. In particular, CD99 and FLI-1 are useful in diagnosing ES/PNET, but these markers can also be expressed in other tumors. CD99 shows diffuse membranous positive staining in virtually all cases of ES/PNET; however, CD99 expression is not specific for ES/PNET because rhabdomyosarcomas, lymphoblastic lymphomas/leukemias, synovial sarcomas, solitary fibrous tumors, and neuroendocrine tumors may also demonstrate CD99 positivity.[7] Positive nuclear staining with FLI-1 is also very sensitive in the diagnosis of ES/PNET, but the FLI-1 antibody will also stain other tumor types, including vascular tumors and lymphoblastic lymphoma.[8] Because positivity for CD99 and/or FLI-1 is suggestive of but not specific for ES/PNET, it is necessary to include them in a comprehensive panel of immunohistochemical markers that will help rule out muscle, lymphoid, epithelial, germ cell, neuroglial, and Merkel cell tumors.
Once ES/PNET is a top consideration in the pathologic differential diagnosis, molecular studies are needed as a complementary diagnostic tool. EWSR1 is one of the most commonly involved genes in sarcoma translocations. ES/PNETs are characterized by specific chromosomal translocations, resulting in a fusion of the EWSR1 gene (22q12) with one of the members of the E26 transformation-specific family of transcription factors.[9] FISH break-apart assay confirmed the presence of a translocation involving EWSR1 in Case 1, but RT-PCR analysis did not detect which specific translocation was present. Several reasons could account for this discrepancy, including sensitivity of PCR to inadequate tissue fixation or insufficient quantity of tumor specimen. Additionally, the tumor from Case 1 may have an EWS-FLI1 exon combination that produces a very long fusion transcript not efficiently amplified from formalin-fixed paraffin-embedded tissue. The patient in Case 2 was 70-years-old, an age group in which ES/PNETs are very uncommon. However, this patient's tumor was found to harbor the EWS-FLI1 translocation, the most common translocation occurring in ES/PNETs. The final diagnosis of ES/PNET in these two cases was made possible by a combination of histological, immunohistochemical, and molecular tests.
MRI is the investigation of choice for the evaluation of lesions within the spinal canal. The tumors described here are primary spinal extraosseous intradural ES/PNETs. On complete imaging workup, there was no evidence of primary lesions elsewhere, to suggest these lesions to be secondary. Primary spinal ES/PNETs are extremely rare, slightly more common in adults than in children and typically located in the region of the filum terminale and cauda equina. Myxopapillary ependymoma is the most common lesion in the region of cauda equina and filum terminale and, therefore, was considered the most likely diagnosis on preoperative imaging. ES/PNET is typically hypointense on T1, hyperintense on T2, and demonstrates heterogeneous enhancement. When associated with hemorrhage, ES/PNET can have hyperintense signal on T1. CSF seeding may produce leptomeningeal enhancement.[10] However, other more common masses including ependymoma, schwannoma, and metastasis show similar imaging findings, making it difficult to differentiate ES/PNET from these lesions.[11] However, if imaging findings of a lesion in the region of the filum terminale and cauda equina are not classical for ependymoma or schwannoma, ES/PNET should be considered, as treatment of this high grade neoplasm is significantly different from the other benign lesions.
CONCLUSION
ES/PNET should be considered in the differential diagnosis of intradural tumors of the spine, despite the low frequency of their occurrence in this location. The diagnostic workup should include imaging, histopathology, immunohistochemical staining, and molecular analysis including both FISH and RT-PCR to identify and characterize the EWSR1 translocation. All data should be considered as a whole when characterizing small round blue cell tumors, in order to arrive at the correct diagnosis and to assure optimal therapy for the patient.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2014/4/1/6/126050
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- A very rare cause of neck pain: Primary Ewing sarcoma of the axis. Pediatr Emerg Care. 2013;29:1197-200.
- [Google Scholar]
- Utility of the immunohistochemical detection of FLI-1 expression in round cell and vascular neoplasm using a monoclonal antibody. Mod Pathol. 2004;17:547-52.
- [Google Scholar]
- Intraspinal Ewing's sarcoma/primitive neuroectodermal tumors. J Clin Neurosci. 2011;18:601-6.
- [Google Scholar]
- A practical approach to the clinical diagnosis of Ewing's sarcoma/primitive neuroectodermal tumour and other small round cell tumours sharing EWS rearrangement using new fluorescence in situ hybridisation probes for EWSR1 on formalin fixed, paraffin wax embedded tissue. J Clin Pathol. 2005;58:1051-6.
- [Google Scholar]
- Soft tissue tumors associated with EWSR1 translocation. Virchows Arch. 2010;456:219-34.
- [Google Scholar]
- Immunohistochemical approaches to the diagnosis of undifferentiated malignant tumors. Ann Diagn Pathol. 2008;12:72-84.
- [Google Scholar]
- Molecular diagnosis in Ewing family tumors: The Rizzoli experience–222 consecutive cases in four years. J Mol Diagn. 2011;13:313-24.
- [Google Scholar]
- Intradural spinal tumours and their mimics: A review of radiolographic features. Postgrad Med J. 2013;89:457-69.
- [Google Scholar]
- From the radiologic pathology archives: Ewing sarcoma family of tumors: Radiologic-pathologic correlation. Radiographics. 2013;33:808-31.
- [Google Scholar]




