
Quadricuspid Aortic Valve: A Rare Congenital Cause of Aortic Insufficiency
Address for correspondence: Dr. Rahul Vasudev, Department of Internal Medicine, St. Joseph's Regional Medical Center, 703 Main Street Paterson, 07503 NJ, USA. E-mail: drrahulvasudev@gmail.com
-
Received: ,
Accepted: ,
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Quadricuspid aortic valve (QAV) is a rare congenital cardiac anomaly causing aortic regurgitation usually in the fifth to sixth decade of life. Earlier, the diagnosis was mostly during postmortem or intraoperative, but now with the advent of better imaging techniques such as transthoracic echocardiography, transesophageal echocardiography (TEE), and cardiac magnetic resonance imaging, more cases are being diagnosed in asymptomatic patients. We present a case of a 39-year-old male who was found to have QAV, with the help of TEE, while undergoing evaluation for a diastolic murmur. The patient was found to have Type B QAV with moderate aortic regurgitation. We also present a brief review of classification, pathophysiology, and embryological basis of this rare congenital anomaly. The importance of diagnosing QAV lies in the fact that majority of these patients will require surgery for aortic regurgitation and close follow-up so that aortic valve replacement/repair is done before the left ventricular decompensation occurs.
Keywords
Aortic regurgitation
congenital valvular diseases
quadricuspid aortic valve
transesophageal echocardiography quadricuspid valves

INTRODUCTION
Quadricuspid aortic valve (QAV) is a rare congenital anomaly with an incidence of 0.01–0.04%.[1] Balinton reported the first known case in 1862.[2] Since then, <200 cases have been reported so far. It is far less frequent as compared to bicuspid (1–2%) or unicuspid aortic valve anomaly. In recent decades, there has been increased awareness of QAV because of better imaging techniques such as transthoracic, transesophageal echocardiography (TEE), and cardiac magnetic resonance imaging (MRI). This increased awareness has contributed to our knowledge of classification, clinical course, and management of this rare congenital condition. More than half of the patients with QAV will require surgical intervention at some point in their life to treat the aortic insufficiency. Hence, early diagnosis and follow-up are critical in these patients. We report a case of asymptomatic QAV diagnosed on TEE.
CASE REPORT
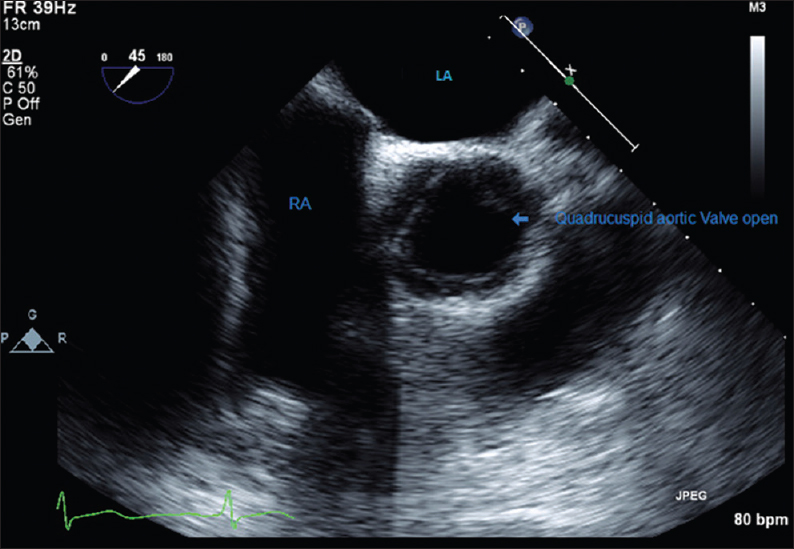
A 39-year-old Hispanic male was seen in his primary care physician's office for follow-up of his hypertension. Patient denied any exertional shortness of breath, chest pain, palpitations, orthopnea, or paroxysmal nocturnal dyspnea. Vitals signs were normal with a blood pressure of 132/58. Cardiac auscultation revealed a diastolic murmur of grade 2/4 in the second right intercostal space. Electrocardiography showed normal sinus rhythm without any abnormalities. Transthoracic echocardiogram was done for the evaluation of this new diastolic murmur, which showed normal left ventricular chamber size with an ejection fraction of 55%. The aortic valve was poorly visualized and showed evidence of mild to moderate aortic insufficiency with valve anatomy suspicious of QAV. There were no other significant abnormalities on echocardiogram. TEE was planned for evaluation of the aortic valve and aortic regurgitation. The aortic valve was found to be quadricuspid [Figure 1 and Video 1] with aortic root measuring 2.40 cm with vena contracta measuring 4.55 mm. There was evidence of moderate aortic insufficiency [Figure 2 and Video 2]. Pulmonary valve was normal with mild pulmonic insufficiency. Tricuspid valve was normal with trace tricuspid regurgitation. The interatrial septum was normal. There was no atrial septal defect or patent foramen ovale. Since the patient was asymptomatic, he was counseled regarding the condition and need for regular follow-up with the cardiologist.

- 39-year-old Hispanic male was examined by his primary care physician for follow-up of his hypertension. Transesophageal echocardiography image shows quadricuspid aortic valve in short axis view, cusp 1, 2, and 3 are equal in size whereas cusp 4 is the accessory cusp and is smaller in size. RVOT: Right ventricle outflow tract; LA: Left atrium; RA: Right atrium.
- 39-year-old Hispanic male was examined by his primary care physician for follow-up of his hypertension. Transesophageal echocardiography with color flow in long axis view shows aortic regurgitation.
DISCUSSION
QAV is a rare congenital anomaly, which is usually diagnosed in the fifth or sixth decade of life when it causes aortic regurgitation. Several different anatomical variations of QAV have been described, Hurwitz and Roberts classified them into seven groups: Type A - four equal cusps; Type B - three equal cusps and one smaller cusp; Type C - two equal larger cusps and two equal smaller cusps; Type D - one large, two intermediate, and one small cusp; Type E - three equal cusps and one larger cusp; Type F - two equal larger cusps and two unequal smaller cusps; and Type G - four unequal cusps.[34] Our patient had Type B quadricuspid with three equal cusps and one smaller cusp as shown in Figure 1. This is the most common type and is often associated with regurgitation due to the unequal shear stress.
Various hypotheses to explain the embryological basis of this anomaly have been presented. One of them is abnormal septation of the embryologic truncus arteriosus. In general, after septation of the arterial trunk, three mesenchymal swellings develop into semilunar leaflets of the aortic and pulmonary trunks. In QAV, however, the fourth cusp arises during the early stage of truncal septation, resulting in either a different number of primordial aortic leaflets or abnormal cusp proliferation.[5] It usually appears as an isolated congenital anomaly but may also be associated with other malformations, the most common being coronary artery anomalies. Development of the aortic valve leaflets occurs just after the development of the coronary artery origins from the sinuses of Valsalva and it is possible that these two groups of anomaly may therefore be embryologically related.[6] It is very important to diagnose these associated anomalies before repair or replacement of aortic valve to avoid ostial obstruction. Aortic dilatation and other structural cardiac abnormalities were relatively common among patients with QAV.[7] In our patient, QAV was not associated with any other congenital anomaly. The physiopathology of the valve dysfunction is poorly understood: Anatomical abnormalities of the cusps could induce unequal shear stress leading to fibrosis and incomplete coaptation.
Although QAV is a congenital malformation, diagnosis is usually late. Before echocardiography was widely used most QAV were diagnosed during surgery or autopsy. Wider use of echocardiography has made detection of QAV easier and hence more frequent.[6] The characteristic echocardiographic finding in short-axis view is an X-shaped commissure pattern during diastole and a rectangular appearance during systole.[8] Transthoracic echocardiography has an important role in detecting congenital anomalies preoperatively. However, transthoracic echocardiography may be suboptimal for recognizing QAV and associated malformations due to the poor acoustic window. TEE provides reliable imaging of heart valves and other structures even in complex congenital heart diseases. Recently, cardiac MRI has also been used for the diagnosis of QAV and other associated congenital cardiac anomalies.
With more liberal use of TEE, computed tomography, and MRI the prevalence of QAV is increasing.[6] The importance of diagnosing this congenital anomaly in asymptomatic adults lies in the fact that more than 50% of these people will require valve repair/replacement in the fifth or sixth decade of life because of worsening aortic regurgitation or stenosis. Repair may be feasible in some patients with regurgitation, but most require replacement.[9] Hence, patients with QAV should have regular follow-up and have aortic valve repair/replacement at an appropriate time before left ventricle decompensation to avoid morbidity, mortality, and complications.
CONCLUSION
Quadricupid aortic valve (QAV) is a rare congenital anomaly. The importance of diagnosing QAV lies in the fact that majority of these patients will require surgery for aortic insufficiency. Once diagnosed patients should be closely followed so that aortic valve replacement/repair is done before the left ventricular decompensation occurs.
Videos Available on: www.clinicalimagingscience.org
Video 1
Video 1 39-year-old Hispanic male was examined by his primary care physician for follow-up of his hypertension. Transesophageal echocardiography shows quadricuspid aortic valve in short axis view Click here to view as Video 1Video 2
Video 2 39-year-old Hispanic male was examined by his primary care physician for follow-up of his hypertension. Aortic regurgitation seen in long axis view of transesophageal echocardiography. Click here to view as Video 1Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2016/6/1/10/179417
REFERENCES
- Incidence, description and functional assessment of isolated quadricuspid aortic valves. Am J Cardiol. 1990;65:937-8.
- [Google Scholar]
- Type F congenital quadricuspid aortic valve: A very rare case diagnosed by 3-dimenional transoesophageal echocardiography. Open Cardiovasc Med J. 2014;8:23-5.
- [Google Scholar]
- Congenital quadricuspid aortic valve associated with aortic insufficiency and mitral regurgitation. J Cardiothorac Surg. 2013;8:87.
- [Google Scholar]
- Quadricuspid aortic valve: Characteristics, associated structural cardiovascular abnormalities, and clinical outcomes. Circulation. 2016;133:312-9.
- [Google Scholar]
- Diagnosis of incompetent quadricuspid aortic valve by two-dimensional echocardiography. Am J Cardiol. 1984;53:972.
- [Google Scholar]
- Outcomes after repair or replacement of dysfunctional quadricuspid aortic valve. J Thorac Cardiovasc Surg. 2015;150:79-82.
- [Google Scholar]




