
Translate this page into:
Multi-level Split Cord Malformation: Do We Need a New Classification?
Address for correspondence: Dr. Gmaan A Alzhrani, Department of Neurosurgery, Montreal Neurological Institution and Hospital, McGill University, Montreal, Quebec, Canada. E-mail: gmaan.al-zhrani@mail.mcgill.ca
-
Received: ,
Accepted: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Split cord malformations (SCMs) are thought to be rare abnormalities representing 3.8-5% of all spinal cord anomalies. The prevalence is estimated to be 1 in 5499 live births (0.02%), with a slight female predominance (1.3:1). Although the estimates of prevalence vary, Type I SCM occurs more frequently than Type II SCM. In this paper, we are reporting the clinical presentation and imaging findings of multi-level SCM in a 27-year-old male. A literature review of the embryological background of SCM and pathological hypothesis for this entity is provided. A systematic review has been conducted to identify multi-level SCM cases reported in the literature, followed by proposing a new classification system to further our understanding and management of SCMs.
Keywords
Multi-level SCM
split cord malformation
spine dysraphism
diastematomyelia

INTRODUCTION

The term split cord malformation (SCM) was first introduced in 1992 by Pang et al., in an attempt to resolve the confusion existing in the pathological definition and the clinical significance of previously existing terminologies in the literature, diastematomyelia and diplomyelia, and the inconsistent usage of these two terms.[1] There are two types of SCM. Type I SCM is the one in which two hemicords are covered by a single dural tube separated by a non-bony fibrous septum. By contrast, Type II SCM differs in that two separate dural tubes hold exclusively the two hemicords associated with an intervening bony spur.[2] In the absence of associated cutaneous stigmata that prompts the clinicians to request screening imaging for the spinal cord, SCM anomalies are usually diagnosed in relatively older children. Although their precise incidence is unknown, SCMs are rare abnormalities representing 3.8–5% of all spinal cord anomalies.[1] In their series, Bademci et al., found only 1 out of 5499 children with primary tethered cord syndrome secondary to diastematomyelia, with an estimated prevalence of 0.02%.[3] Similar to the other occult spinal dysraphisms, there is a slight female predominance (1.3:1).[3] Although the estimate of prevalence may vary, Type I SCM occurs more frequently than Type II SCM.[4] In a study conducted by Mahapatra, Type I SCM was seen in 156 patients (61.4%) and Type II SCM in 98 patients (38.6%).[5] Both types of SCM can occur in open spine dysraphism as in myelomeningocele.[6]
Regarding the localization of SCM, a recent retrospective study reported that the thoracic spinal cord was the most common site for SCMs, accounting for 38.8% of cases, followed by lumbar region in 28.6%, thoracolumbar in 22.4%, cervicothoracic region in 6.1%, and lumbosacral region in 4.1%.
SCM is usually associated with other congenital anomalies. The most common association is tethered cord, scoliosis or kyphoscoliosis, syringomyelia, and spina bifida.[15] However, SCM may coexist with myelomeningocele in 15-39% of the cases,[46] and it is more likely to be Type I which tends to occur at or just rostral to the level of myelosmeningocele defect.[14]
In most of these reports, the SCMs affect a single metameric level of the spinal cord. The occurrence of multi-level SCM is a rare event. Here, we report a young male with this condition and propose an additional modification to the classification system of SCM to reflect the clinical significance associated with such a finding.
CASE REPORT
A 27-year-old male presented to our clinic complaining of chronic low back pain for a year and half after he sustained minor back trauma at work. His pain was radiating to both lower extremities, but was more to the right side, for which he underwent many physiotherapy sessions. However, despite this, his radiculopathy persisted. Physical examination revealed slight weakness of plantar flexion in the right foot and mild decreased sensation of the right L5-S1 dermatomes. Magnetic resonance imaging (MRI) of his spine showed an arachnoid cyst posterior to T5 and T6 segments of the spinal cord causing anterior displacement of the cord and posterior cord flattening [Figure 1]. Starting at the level of T7-T8, splitting of the spinal cord was observed and continued down to the level of T12 where the two hemicords then fused back to a normal conus medullaris [Figure 2]. The low-lying conus medullaris that once again split into two hemicords at the L3-L4 level with a low-lying tethered cord inserted posteriorly at S1 is seen in Figure 3.
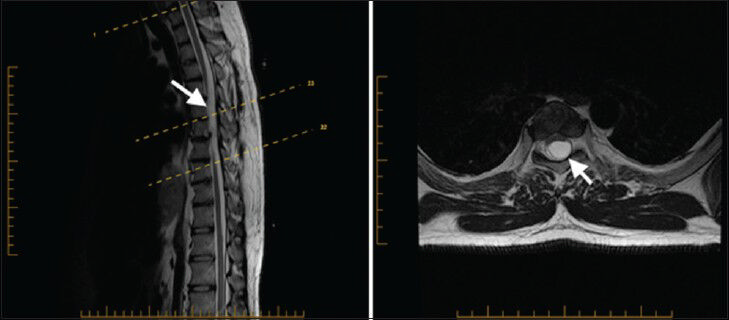
- 27-year-old male presented with chronic low back pain for a year and half after he sustained minor back trauma at work. Pain was radiating to both lower extremities, but more to the right side. It was later diagnosed as a form of spinal cord malformation. a) Sagittal T2W and b) axial T2W MRI of the thoracic spine show arachnoid cyst posterior to T5 and T6 segments of the spinal cord causing anterior displacement of the cord and posterior cord flattening (arrowhead). Epidural arachnoid cyst herniation through the posterior dura is a possible differential diagnosis.
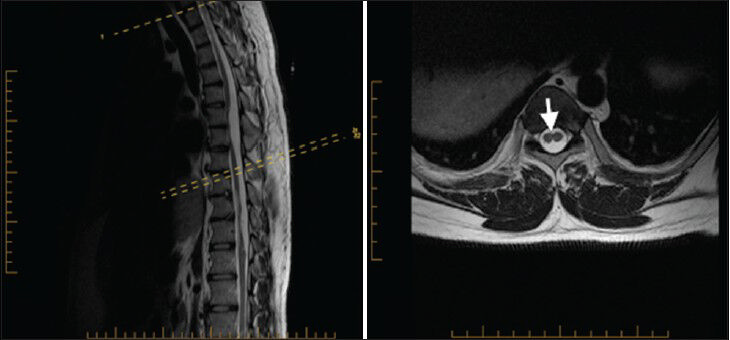
- 27-year-old male presented with chronic low back pain for a year and half after he sustained minor back trauma at work. Pain was radiating to both lower extremities, but more to the right side. It was later diagnosed as a form of spinal cord malformation. a) Sagittal T2W and b) axial T2W MRI of the thoracic spine show split cord (arrowhead) starting at the level of T7–T8 spine segment and continuing down to the level of T12 where the two hemicords then fused back into normal conus medullaris.
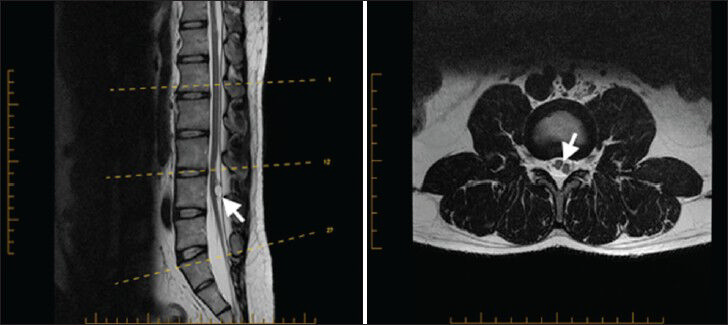
- 27-year-old male presented with chronic low back pain for a year and half after he sustained minor back trauma at work. Pain was radiating to both lower extremities, but more to the right side. It was later diagnosed as a form of spinal cord malformation. a) Sagittal T2W and b) axial T2W MRI of the lumbar spine show a low-lying conus medullaris that once again split into two hemicords (arrowhead in b) at the L3–L4 level, with a low-lying tethered cord inserted posteriorly at S1. Notice in the sagittal T2W MRI the presence of intradural hyperintense circular lesion and hyperintense lesion in T1W (not shown) characteristic for lipoma (arrowhead in a).
DISCUSSION
In normal human embryonic development, the continuous proliferation of cells during the first 2 weeks form a hollow spherical structure called blastocyst that is surrounded by the cell layers of the trophoblast. The blastocysts located eccentrically within this sphere form two cellular layers, epiblast facing the amniotic cavity dorsally, and hypoblast facing the yolk sac cavity ventrally. The transformation of a two-layer embryo into a three-layer embryo (endoderm, mesoderm, and ectoderm) is termed gastrulation process. During this process an ingression of the epiblast through the primitive groove toward the primitive streak occurs which forms at the caudal end of the embryo. Mesodermal cell layer migrates through the primitive pit, which is situated at the cranial end of the primitive groove, to become the future notochordal process. By post-ovulatory day 16, the primitive streak starts to regress as the notochordal process elongates and by days 18-20, forms a canal termed the notochordal canal, which communicates dorsally with the amniotic cavity and becomes open to the yolk sac ventrally through a process known as intercalation. The notochordal process becomes the primitive neuroenteric canal. After 2-8 days of formation of the primitive neuroenteric canal, it separates from the endoderm by a process known as extracalation.[1] After that, the canal obliterates and a true notochordal rod forms above which the neural tube forms during the primary neurulation. The closure of the neural tube is an uniform process that proceeds in a craniocaudal fashion at its dorsal aspect, starting at separate distinct initiating closure points, leading to sealing of the cranial neuropore on days 23-25 and the caudal neuropore on days 25-27 of embryonic life.[1] Failure of the intercalation and persistent primitive neuroenteric canal will result in inhibition of midline dorsal fusion of the neural tube and subsequently to a split cord. Because the formation of the primitive neuroenteric canal occurs in caudal–cranial direction from the primitive pit, the split cord must be rostral to the canal.[1] The neuroenteric canal subsequently gets invested with the mesenchymal cells to form an endomeseynchmal cleft between the notochord and the ectodermal layer splitting the neural tube. According to the unified theory proposed by Pang in 1992, the development of the intervening structure (bony spur vs. fibrous septum) depends on which way the mesenchymal cells differentiate to form bone or cartilage.
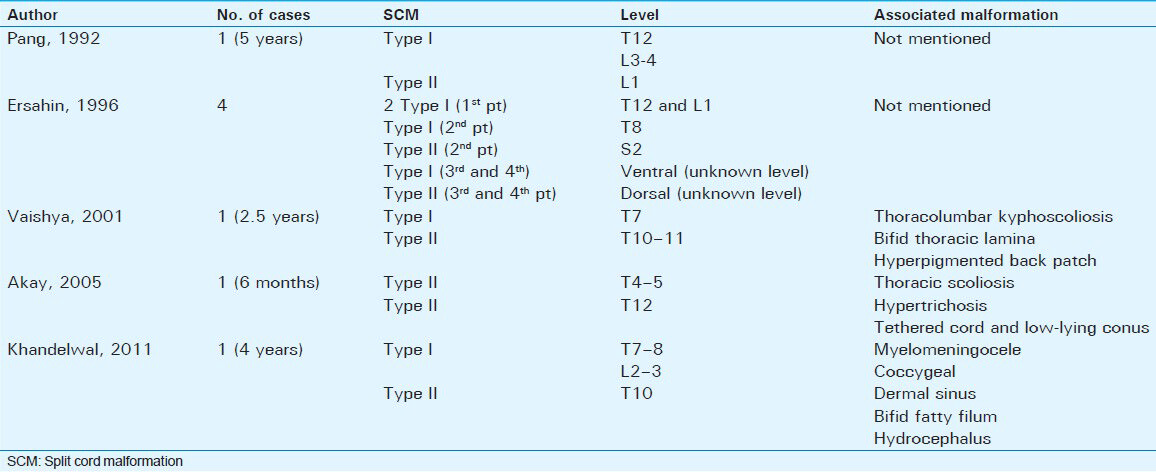
Composite SCMs are rare, accounting for less than 1% of all SCMs.[7] A review of the English literature revealed only eight cases diagnosed with composite SCM [Table 1]. The majority of these cases presented during childhood and either thoracic or lumber spine was involved in these cases, except for one case where the author reported Type II SCM at the second sacral vertebra without mentioning how low the tethered cord was,[7] raising some suspicion regarding the diagnosis in that case. The previously mentioned cases showed that despite having a clear definition for Type I and Type II SCM, we still have some confusion in the diagnosis of these cases with multiple split points in the cord and we believe this could necessitate adding an additional Type III SCM. Pang[1] reported a 5-year-old girl with Type I SCM at T12 and L3-L4 level and Type II at L1 level, but did not mention any associated anomalies. Ersahin[7] reported four cases: One patient with two tandem Type I SCM at T12 and L1, the second patient with Type I SCM at T8 and Type II SCM at S2, and the third and fourth patients with Type I SCM ventrally and Type II SCM dorsally at the same level. Vaishya[8] reported one patient with Type I SCM at T7 and Type II SCM at T10-T11, associated with thoracic and thoracolumbar kyphoscoliosis. Akay[9] reported one patient with tandem Type II SCM at T4-T5 and T12, associated with thoracic scoliosis, tethered low-lying conus, and hypertrichiosis. Khandelwal[10] reported a patient with Type I SCM at T7-8 and L2-3 and Type II at T10 level, associated with myelomeningocele, coccygeal dermal sinus, bifid fatty filum, and hydrocephalus.
Pang, in his reported case, proposed a single large endomesenchymal tract underlying the multiple non-contagious split cord segments. However, there was no interim normalization of the cord. Therefore, he claimed that the presence of multiple accessory neuroenteric fistulas (endomesenchymal tract) in this multi-segmental SCM is exceedingly rare,[1] and we believe it carries more significant morbidity as reported by Khandelwal.[10]
Our patient presented in adulthood unlike the reported cases, although he had mild right side plantar flexion weakness and decreased sensation over the right L4-L5 dermatome which he attributed to the trauma he had in the past. It is unclear to us if this neurological deficit was present since childhood and probably was not preventing him from doing his usual physical activity, as he did not seek medical attention for that until he got the trauma. On the other hand, late presentation of SCM during adulthood has been attributed to many participating factors like trauma, vigorous exercises involving flexion of the hip joints and the spinal column, spinal canal narrowing, and the presence of associated congenital anomalies in the local area. All these factors can cause stretching of the preexisting tethered cord and percipitate neuronal injury. Our patient had loculated cerebrospinal fluid (CSF) collection (arachnoid cyst) behind the T5 and T6 segments of the cord [Figure 1]. This caused marked anterior displacement of the cord and posterior cord flattening. Just below this level, there was a diastematomyelia present, which continued down to the T12 level, where the two hemicords then fused back into normal conus medullaris. The conus medullaris then once again split into two hemicords at the L3-L4 level in what appears to be a low-lying tethered cord. Finally, there was an ovoid-shaped spontaneously hyperintense lesion behind the two hemicords at the level of L4. This was probably a fat-containing lesion (lipoma), and the distal most aspect of the tethered cord inserted posteriorly at S1.
Based on this review, we believe there should be a distinct type of SCM to classify multiple non-contagious SCMs. We propose Type III multi-level SCM with three subdivisions: Type IIIA, with a bony spur at both levels, Type IIIB, with a fibrous septum at both levels, and Type IIIC, with mixed fibrous and bony spur or associated SCM anomalies [Table 2]. This is a descriptive anatomical–pathological modification of the original Pang's classification put forward in order to help improve our understanding of future cases, as little is known about the natural history of this particular type of SCM.

CONCLUSION
Multi-level non-contagious SCMs are very rare entities with only eight reported cases in the literature. In this paper, we have provided a systematic review of all the reported cases, along with an anatomical and pathological description of the malformation. We proposed adding a new classification to further our understanding of the natural history of SCMs and the consequences of surgical interventions.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2014/4/1/32/135181
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- Split cord malformation: Part I: A unified theory of embryogenesis for double spinal cord malformations. Neurosurgery. 1992;31:451-80.
- [Google Scholar]
- Split spinal cord malformation: Report of 5 cases in a single chinese center and review of the literature. Pediatr Neurosurg. 2013;49:69-74.
- [Google Scholar]
- Prevalence of primary tethered cord syndrome associated with occult spinal dysraphism in primary school children in Turkey. Pediatr Neurosurg. 2006;42:4-13.
- [Google Scholar]
- Split cord malformation: Part II: Clinical syndrome. Neurosurgery. 1992;31:481-500.
- [Google Scholar]
- Split cord malformations: A clinical study of 254 patients and a proposal for a new clinical-imaging classification. J Neurosurg. 2005;103(Suppl 6):531-6.
- [Google Scholar]
- Occurrence of split cord malformation in meningomyelocele: Complex spina bifida. Pediatr Neurosurg. 2002;36:119-27.
- [Google Scholar]
- Split cord malformations: Report of three unusual cases. Pediatr Neurosurg. 1996;24:155-9.
- [Google Scholar]
- Split cord malformation: Three unusual cases of composite split cord malformation. Childs Nerv Syst. 2001;17:528-30.
- [Google Scholar]
- Composite type of split cord malformation: Two different types at three different levels: Case report. J Neurosurg. 2005;102(Suppl 4):436-8.
- [Google Scholar]
- An unusual case of 4 level spinal dysraphism: Multiple composite type 1 and type 2 split cord malformation, dorsal myelocystocele and hydrocephalous. J Pediatr Neurosci. 2011;6:58-61.
- [Google Scholar]




