
Translate this page into:
Congenital Pulmonary Artery Anomalies: A Review and Approach to Classification
Address for correspondence: Dr. Priya G Sharma, Department of Radiology, Division of Pediatric Imaging, University of Florida College of Medicine, Gainesville, FL 32608, USA. E-mail: sharpg@radiology.ufl.edu
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Congenital pulmonary artery anomalies are infrequent but given improved prenatal diagnosis and care, and neonatal surgical advances, over the past two decades are not uncommonly encountered by cardiothoracic imagers. An understanding of their etiology, classifications, associated anomalies, and surgical management can be helpful to avoid under or overdiagnosis. Timely diagnosis assisted by familiarity with imaging findings across modalities and recognition of surgical findings allows for medical management and surgical planning for these patients, with more patients reaching adulthood than ever before.
Keywords
Anomalous origin of a branch pulmonary artery from ascending aorta
left pulmonary artery sling
main pulmonary artery
right pulmonary artery
unilateral pulmonary artery agenesis


INTRODUCTION
Advances in imaging and clinical care have resulted in greater identification of patients with congenital pulmonary artery (PA) anomalies and have raised the need for an updated review on the spectrum of these pathologies.
We provide an imaging review, along with brief common clinical presentations and surgical options for treatment, which can be helpful for both general and pediatric radiologists.
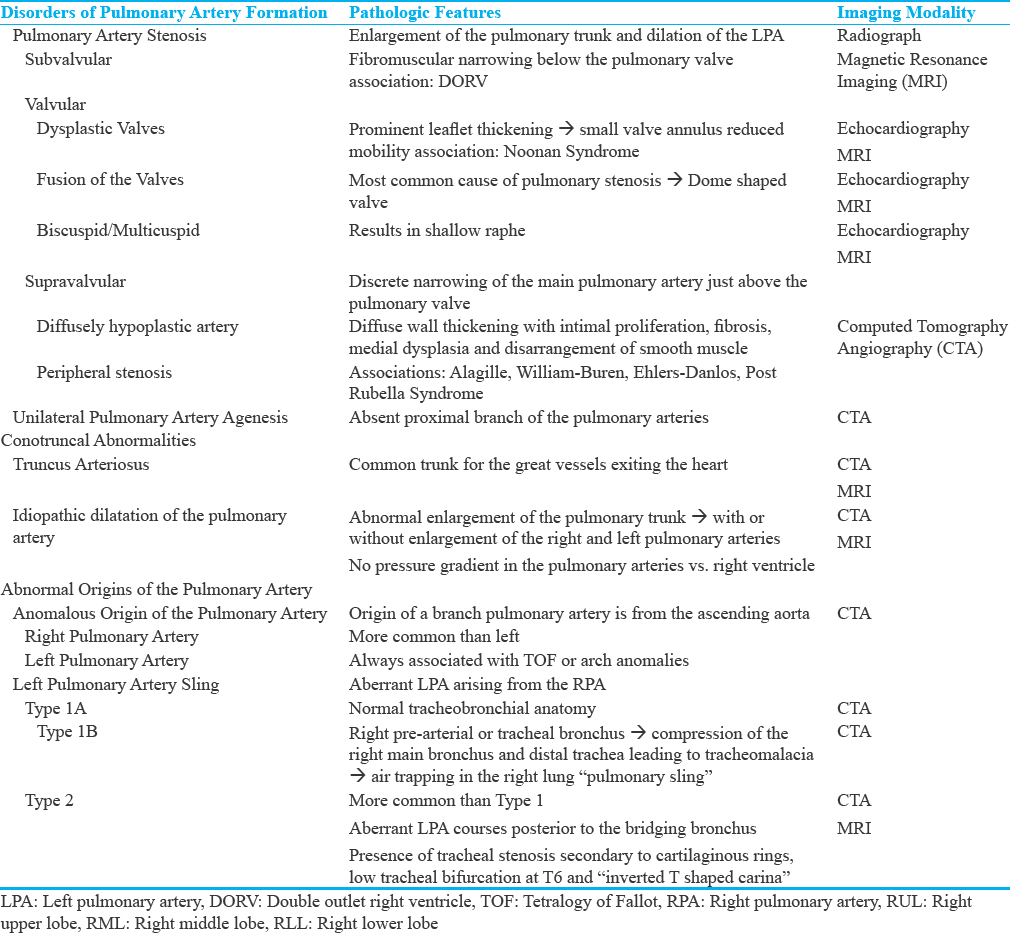
Congenital PA anomalies have a variable presentation which is often dependent on the underlying nature of the vascular abnormality. Although a classification scheme has not been addressed in the literature thus far, a spectrum of findings related to the formation of the pulmonary arteries can be proposed to subdivide these anomalies [Table 1]. Disorders of PA formation may result in disorders of pulmonary stenosis or unilateral PA agenesis (UPAA). Entities such as truncus arteriosus and idiopathic dilatation of the PA (IDPA) are felt to arise from failure during conotruncal embryogenesis. Anomalous origin of the PA and left PA sling (LPAS) are characterized by aberrancy in the origin of the PA.
IMAGING OF PEDIATRIC CARDIAC EXAMINATIONS AT OUR INSTITUTION
Pediatric computed tomography angiograms (CTA) at our instruction are chiefly performed on a 320-detector row scanner (Acquilion One, Cannon Medical Systems). The examination is performed with slice thickness of 0.5 mm at 0.25 mm interval. The examination is performed with dose reduction technologies including Target CTA (Cannon Medical Systems) with which the entire heart is imaged with prospective. Electrocardiogram gating in one rotation along with dose modulation (SUREexposure, Cannon Medical Systems). Contrast for the examination is calculated by weight at 1.43 ml/kg. For patients under 49 kg, this is given through rapid hand injection. For patients over 49 kg, the contrast is power injected intravenously at 3 ml/second for two-thirds of the contrast bolus volume, followed by 50% contrast to 50% saline dilution at 2 ml/second for the remaining volume. The examinations are gated with conversion of heart rate per minute to milliseconds between R to R interval, which is then used to determine the exposure window and target percent [Table 2].

Pediatric magnetic resonance imaging (MRI) examinations can be particularly challenging in young patients due to their inability to tolerate the time it takes to perform the examination. When necessary, we attempt to avoid anesthesia and opt for gated CTA examination. At our institution, pediatric MRI examinations requiring sedation are performed in-house on 3 Tesla magnet (Vantage, Titan, Cannon Medical systems) with anesthesia monitoring. Acquisition sequences include three plane localizers, followed by axial and angled with the arch echo-planar fast spin echo sequence (HASTE); two, three, and four chamber cine images; short-axis cine images; and coronal angiogram sequences. Additional sequences are added at the discretion of the radiologist, such as flow quantification sequences and MR angiogram and venogram sequences. This protocol is adjusted during patient scanning by the interpreting physician at the worksite.
PEDIATRIC CARDIAC IMAGING DOSE REDUCTION
Radiation dose awareness is central in imaging pediatric patients with congenital PA anomalies. However, diagnostic quality of imaging is also paramount. As CT does provide better spatial resolution than MRI, this modality is often favored for imaging complex congenital heart disease requiring surgical intervention. Furthermore, CT examinations can be performed using low-dose techniques with overall low effective doses.[1] Utilizing low-dose technique is important in these patients who may require multiple imaging studies through their disease course. At our institution, a typical-gated CTA examination in a neonate results in a dose–length product of approximately 20 mGy cm.
MRI does not subject the patient to any ionizing radiation and is more useful in cases where there is a concern about blood flow, shunting, and/or pressure gradients. However, cardiac MRI of patients under the age of 8 at our institution does require anesthesia. Our institution serves as a tertiary referral center and CT is more readily and easily available. MR, as it requires sedation in younger patients is less readily available given the confines of the pediatric anesthesiologists and scheduling constraints. Often these patients travel to our tertiary care center for same-day cardiology clinic appointments after which, because of its speed and availability, same-day CTA is often a more convenient choice.
PULMONARY ARTERY SIZE MEASUREMENTS
Both the main PA (MPA) diameter and ratio of the MPA to the ascending aorta (AA) measurements have been shown to be easily obtained and reproducible.[2] Measurement of the PA is traditionally performed at the level of the bifurcation of the right PA (RPA) and the AA measurement is obtained at the same imaging plane to obtain a ratio. The Framingham Heart Study cutoff values for adults have been suggested for the MPA for men as 29 mm and for women as 27 mm. The MPA to AA ratio cutoff of 0.9 has been suggested for both men and women.
At our institution, for pediatric patients, we use Z-scores which are based on nomograms based on body surface area and gender to determine whether the PA is dilated.[3]
DISORDERS OF PULMONARY ARTERY FORMATION
Pulmonary artery stenosis
PA stenosis is defined as the obstruction of flow from the right ventricle to the pulmonary arteries. This typically occurs at the level of the pulmonary valve but may occur anywhere from the level of the pulmonary valve to the peripheral PA branches. During the 5th week of gestation, the bulbus cordis separates into the AA and the MPA. The pulmonary valves develop from a section of the bulbus cordis and moves from a location that is posterior to the aortic valve to one that is anterior and to the left of the aortic valve by the end of the 5th week of gestation. It is thought that maldevelopment of the distal portion of the bulbus cordis leads to valvular pulmonary stenosis.[4] The most common level of PA stenosis occurs at the level of the pulmonary valve and is typically secondary to fusion or absence of the commissures with thickened pulmonary valve leaflets. This results in a dome-shaped pulmonary valve and leads to obstruction of flow during systole and accounts for 40%–60% of congenital pulmonary valve stenosis.[5]
Congenital pulmonary stenosis is often asymptomatic when diagnosed. The most common finding on radiograph is enlargement of the pulmonary trunk and left PA (LPA), which is due to poststenotic dilatation [Figure 1]. Dilation of the LPA is seen as a result of the “jet stream” effect, but the degree of dilatation is not related to the severity of obstruction.[6]
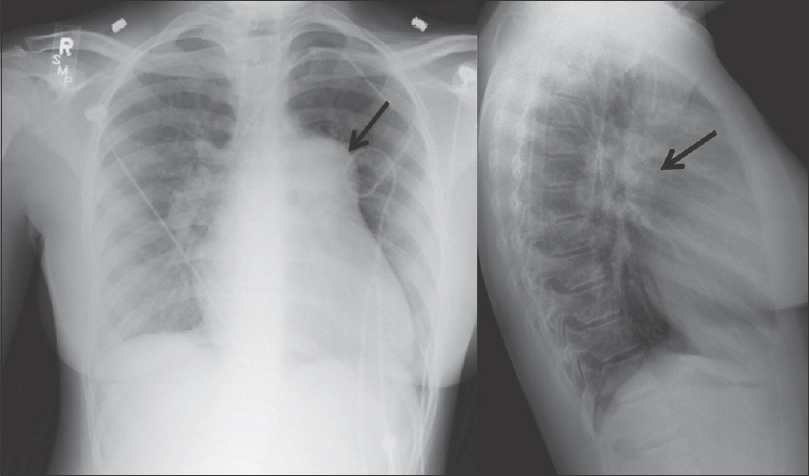
- A 39-year-old female with a history of congenital heart disease, which included atrial septal defect and right pulmonary artery stenosis. The patient underwent atrial septal defect repair and angioplasty of the pulmonary artery. Pulmonary artery and lateral radiographs of the chest show enlargement of the pulmonary trunk and left pulmonary artery (black arrow) in a patient with congenital pulmonary artery stenosis.
Although pulmonary stenosis may be an isolated finding, it is often syndromic and/or associated with cardiac abnormalities. A form of valvar pulmonary stenosis with dysplastic pulmonary valves is often associated with Noonan Syndrome and is characterized by irregular valves with prominent leaflet thickening resulting in a small valve annulus and reduced mobility.[7] Other variants seen at the valvular level include bicuspid or multi-cuspid valves. In the setting of bicuspid valve, one leaflet may be larger than the others, with a shallow raphe or both leaflets may be the same size.[8] Patients who have quadricuspid pulmonary valves are more likely to be asymptomatic. However, it is very important to diagnose patients before undergoing the Ross Procedure, pulmonary autografting, as this may result in valve dysfunction.[8]
Subvalvular pulmonary stenosis is caused by fibromuscular narrowing below the pulmonary valve and is often seen in association with a double-chambered right ventricle.[9] This uncommon condition can be dynamic as well, with increased restriction of blood flow with the right ventricular contraction. In contrast, the subvalvular stenosis that is seen in Tetralogy of Fallot is more fibrotic and shows little change during systole and diastole.[6]
Supravalvular pulmonary stenosis occurs when there is a discrete narrowing of the MPA – just above the pulmonary valve. This stenosis can vary from a thin diaphragm to a diffusely hypoplastic artery and is further characterized as peripheral PA stenosis when there is unilateral or bilateral distal narrowing in the more distal PAs. The peripheral variant is usually associated with syndromes such as Alagille, William–Beuren, Ehlers–Danlos, and the postrubella syndrome. The pathological process is diffuse wall thickening with intimal proliferation, fibrosis, medial dysplasia with hypertrophy, and a nonparallel arrangement of smooth muscles.[10] These stenoses can have a varied appearance from discrete areas of narrowing on CT to long diffuse hypoplastic segments and are best characterized on CT angiography [Figure 2].
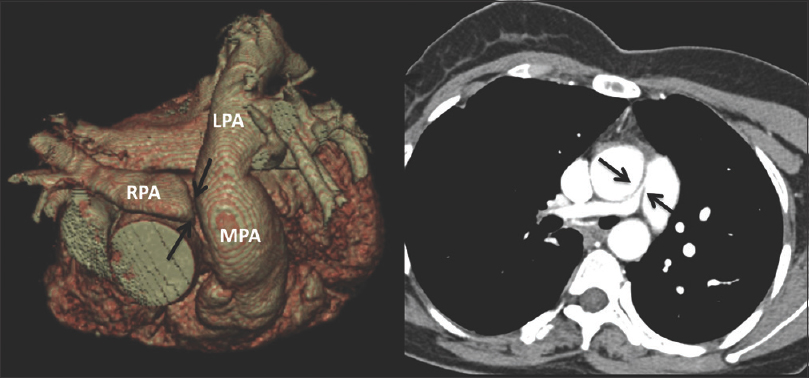
- The same patient as in Figure 1. 3D reconstruction and axial computed tomography angiogram shows severe short-segment discrete narrowing of the proximal right pulmonary artery (black arrows).
Treatment for PA stenosis is based on the location and degree of narrowing. For patients with valvular disease balloon valvuloplasty is the first line of treatment.[11] Balloon valvuloplasty is also the favored treatment in the neonatal stage with critically defined pulmonary stenosis.[11] Patients are concomitantly often treated with prostaglandin to encourage the right-to-left shunting. Surgical treatment is reserved for those patients with dysplastic pulmonary valves or hypoplastic MPA. Muscular resection is additionally often required in these patients.[12]
Unilateral pulmonary artery agenesis
In UPAA one of the proximal branch pulmonary arteries is absent despite normal development of the intrapulmonary vessels.[1314] The UPAA may be isolated or seen in association with congenital heart disease cases in <1% of cases, which include right-sided aortic arch, septal defects, truncus arteriosus, and tetralogy of Fallot. In neonates, the distal intrapulmonary vessels are usually supplied by a patent ductus arteriosus. In the rare case of bilateral PA discontinuity, the pulmonary blood supply is completely dependent on the patency of the ducti arteriosi.[13]
UPAA is often asymptomatic during early childhood. When symptoms do develop, they include recurrent pneumonia, hemoptysis, exercise intolerance, and pulmonary hypertension of the contralateral lung.[15] Children with underlying congenital heart disease often present much earlier secondary to cardiac symptoms, at which time the PA anomaly is incidentally found.
Embryogenesis of unilateral PA discontinuity is controversial. Nonetheless, there is a predominating theory suggesting involution of one of the proximal sixth aortic arches leading to failure of the ipsilateral branch PA to form. However, if the ipsilateral distal sixth aortic arch persists, it will instead join the ipsilateral lung bud, thereby connecting the ductus arteriosus directly to the intrapulmonary arteries.[15] If this anomaly is not repaired in the neonatal period, the blood flow to the affected lung will be diminished on ductus arteriosus closure and the intrapulmonary portion of the artery becomes hypoplastic, in which case surgical repair will be less likely to be effective.[15]
Chest radiography in UPAA may demonstrate mediastinal shift toward the affected side or decrease in pulmonary vascular markings in the affected lung. Ventilation-perfusion scanning will demonstrate normal ventilation but the complete absence of perfusion to the affected lung. The utility of echocardiography is limited in this diagnosis, but secondary signs of right ventricular hypertrophy and a dilated MPA may be seen. Cardiac catheterization and pulmonary angiography will show a hypoplastic proximal branch PA. An ipsilateral pulmonary vein wedge venogram may demonstrate retrograde filling of the normal intrapulmonary arteries. CT angiogram offers a noninvasive method to depict UPAA and can demonstrate any collateral vessels that may be present [Figures 3 and 4].
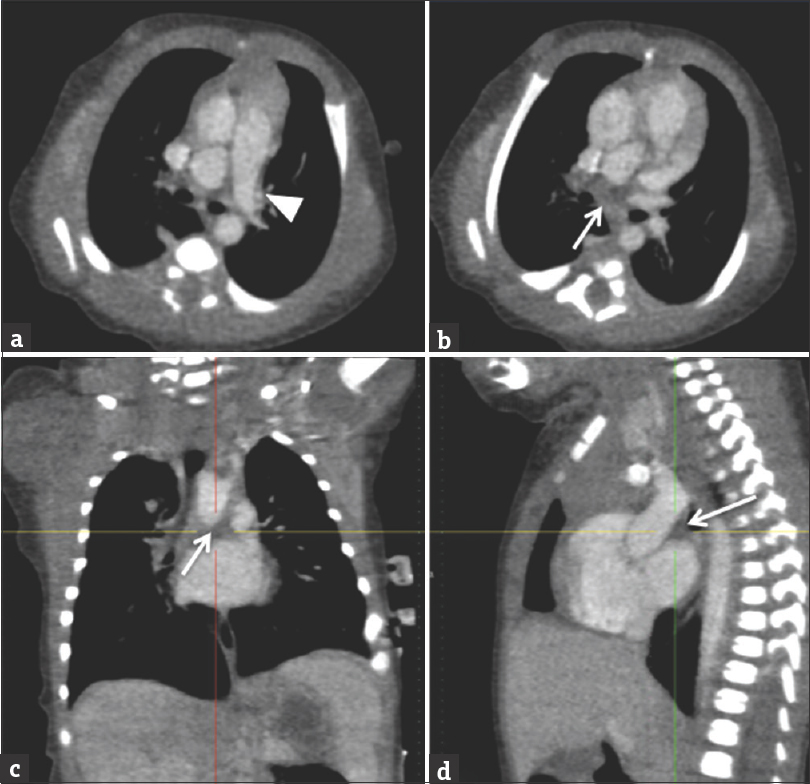
- A 3-day-old female with a history of cardiac abnormalities and symptomatic respiratory distress. Computed tomography angiogram images (a and b) are selected axial images, (c) is a coronal reconstruction, and (d) is a sagittal reconstruction. The main and left pulmonary artery opacify with contrast (white arrowhead). The right pulmonary artery is absent (white arrows demonstrate expected location). No large major aortopulmonary collateral arteries from the aorta or major branch vessel was identified in this patient. There is an incidental azygous lobe noted in the right lung.
![Computed tomography angiogram [same patient as Figure 3] demonstrating poor contrast opacification of the distal right pulmonary artery in the right lower lobe (white arrow). The right pulmonary veins are also small in caliber (white arrowhead).](/content/12/2018/8/1/img/JCIS-8-29-g007.png)
- Computed tomography angiogram [same patient as Figure 3] demonstrating poor contrast opacification of the distal right pulmonary artery in the right lower lobe (white arrow). The right pulmonary veins are also small in caliber (white arrowhead).
Early surgical repair of UPAA is desirable as the patent ductus arteriosus often regresses by 6–12 months of age and the intrapulmonary arteries then become hypoplastic, limiting surgical options and long-term outcomes.[15] Embolization and pneumonectomy may be required in some cases due to recurrent bleeding or bronchopneumonia.
CONOTRUNCAL ABNORMALITIES
Truncus arteriosus
Truncus arteriosus accounts for 1% of congenital cardiac disease and is defined by a single great artery supplying the systemic, coronary, and pulmonary blood flow. The single truncal valve may have two to five leaflets, which may be stenotic or insufficient. An atretic pulmonary valve and a ventricular septal defect (VSD) may also be present.[16] Truncus arteriosus is rare with incidence of in 1 in 10,000 births and approximately 32% of cases are associated with a 22q11.2 deletion and DiGeorge syndrome.
In normal embryology, the conotruncus splits into the aorta and PA trunks. When there is partial or complete failure of this separation, there is a common trunk for the great vessels exiting the heart resulting in a truncus arteriosus. Typically, neonates are initially asymptomatic while the PA pressures are elevated. However, they typically present over the first few weeks of life with congestive heart failure due to the persistently increased PA flow. If the anomaly is not repaired by 3–6 months, these infants eventually develop PA hypertension which may become so severe that retrograde flow during diastole can create a steal phenomenon from the abdominal vasculature and increase the risk of necrotizing enterocolitis.[17]
A chest radiograph is frequently the initial imaging modality obtained for a cyanotic neonate and may show a right aortic arch, cardiomegaly, and possibly increased pulmonary vascular markings. Although a radiograph is the first imaging modality, an echocardiogram better demonstrates the anatomy of the truncus, aortic arch, and origin of the pulmonary arteries. In addition, the presence and size of a VSD and function of the truncal valve can be well evaluated on echocardiogram. Imaging with CT or MR aids in identification of additional anomalies in the aortic arch and provide superior visualization of the venous connections of the heart compared to echocardiogram [Figures 5–7]. The coronary artery anatomy can also be evaluated, which is essential for surgical planning and may negate the need for cardiac catheterization.[18]
![A 5-day-old term infant with prenatal concern for truncal abnormality. Axial images [Figure 5a to c] from a cardiac computed tomography angiogram demonstrating Type I truncus arteriosus with arch interruption. The aorta (black arrow) and main pulmonary artery (white arrow) are shown to arise from a common trunk (black arrowhead). The branch pulmonary arteries both arise from the posterior aspect of the common trunk (white arrows).](/content/12/2018/8/1/img/JCIS-8-29-g008.png)
- A 5-day-old term infant with prenatal concern for truncal abnormality. Axial images [Figure 5a to c] from a cardiac computed tomography angiogram demonstrating Type I truncus arteriosus with arch interruption. The aorta (black arrow) and main pulmonary artery (white arrow) are shown to arise from a common trunk (black arrowhead). The branch pulmonary arteries both arise from the posterior aspect of the common trunk (white arrows).
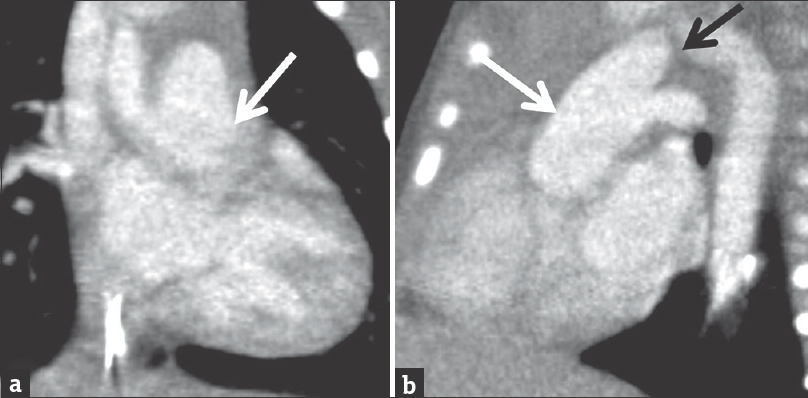
- Coronal (a) and sagittal (b) reconstructions from the same computed tomography angiogram of the chest presented in Figure 5. The common trunk of aorta and main pulmonary artery (white arrow) are demonstrated as well as interruption of the aortic arch proximal to the origin of the left subclavian artery (black arrow).
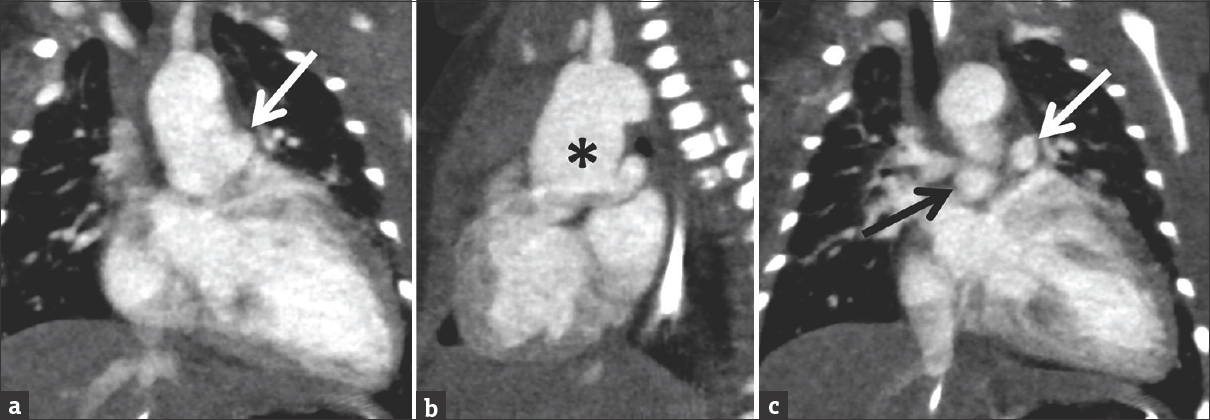
- Coronal images from a cardiac computed tomography angiogram in a 7-day-old male with Type II truncus arteriosus which was suspected on echocardiography. There is a dilated ascending aorta (black asterisk) with the branch pulmonary arteries arising separately from the posterior wall of the ascending aorta (right pulmonary artery = black arrow; left pulmonary artery = white arrow).
Surgical repair of a truncus arteriosus with a Rastelli procedure is desired by 3 months of age to prevent PA obstruction. The prognosis for truncus arteriosus is poor if not repaired, with a 90% mortality rate by 1 year of age as compared to 5% for those patients who have undergone repair.[19]
Idiopathic dilatation of the pulmonary artery
IDPA is a rare congenital abnormality whose natural occurrence remains unknown. It is characterized by abnormal enlargement of the pulmonary trunk, with or without enlargement of the right and left pulmonary arteries.[20] Patients affected by this condition are usually asymptomatic. It is a diagnosis of exclusion. The primary differential considerations include pulmonary stenosis and pulmonary arterial hypertension. In the setting of pulmonary arterial hypertension, there is elevation of the pressures in the pulmonary arteries and the right ventricle. In IDPA, there is no pressure gradient.
The etiology of this disease is unknown although an unequal division of the truncus arteriosus has been suggested, as has congenital weakness of the arterial wall. Connective tissue diseases have also been implicated in PA dilatation.
On chest X-ray convex, opacity in the left aspect of the mediastinum is seen, representing the enlarged pulmonary trunk. Thoracic CT and MRI are useful in demonstrating severe expansion of the PA. Furthermore, CT may be useful in early detection of aneurysmal wall changes that could suggest impending rupture [Figure 8].[21] In younger patients, MRI is favored to reduce radiation exposure [Figure 9]. As most of these patients are asymptomatic, conservative medical management is often used. However, when the PA reaches a diameter of 60 mm or more, surgical repair is considered.[22]
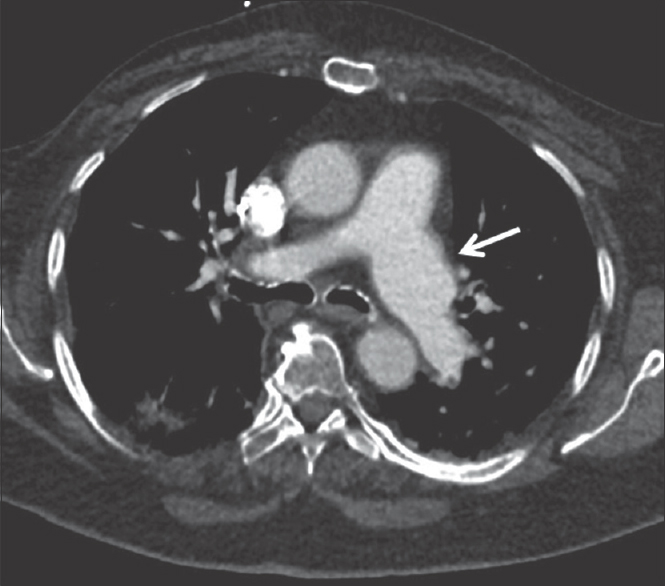
- A 31-year-old patient with suspected connective tissue disorder. Axial computed tomography angiogram image from a young adult patient shows idiopathic dilatation of the left pulmonary artery (white arrow) with normal caliber main and right pulmonary artery.
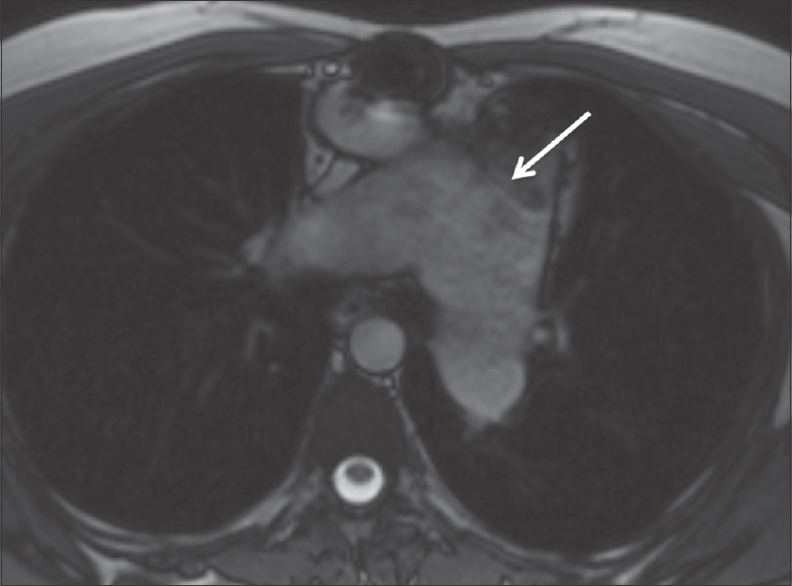
- Axial true fast imaging with steady-state free precession from the same patient in Figure 8 showing dilated main pulmonary artery (white arrow) which measured up to 6.4 cm in diameter. The branch pulmonary arteries are also dilated.
ABNORMAL ORIGINS OF THE PULMONARY ARTERY
Anomalous origin of the pulmonary artery
Anomalous origin of a branch PA from AA (AOPA)/hemitruncus accounts for 0.3% of congenital heart disease. The RPA is more commonly involved than the LPA.[23] Cases of left-sided AOPA are always associated with tetralogy of Fallot and/or aortic arch anomalies, whereas right-sided AOPA is more commonly associated with a patent ductus arteriosus or aortopulmonary septal defect.[23]
Separate aortic and pulmonic valves are a distinguishing characteristic from the shared single truncal valve seen in truncus arteriosus. The aortic origin of a branch PA creates a large left-to-right shunt leading to pulmonary hypertension and eventually pulmonary edema. Pathogenesis of AOPA is complex with potential different embryological causes for proximal anomalous RPA, distal anomalous RPA, and anomalous LPA. On the right, if there is incomplete migration of the right sixth aortic arch to the left side, a proximal right-sided AOPA will result. The origin for a more distal right-sided AOPA is less well understood and may be related to the development of a right fifth aortic arch and absence or early involution of the distal right sixth aortic arch.[23] On the left, there is thought to be failure of the left sixth arch to develop preventing fusion of the LPA to the MPA, similar to the proposed pathogenesis for a distal right-sided AOPA.
On chest radiograph, there are increased pulmonary vascular markings in the lung ipsilateral to the AOPA, cardiomegaly, and possibly a right-sided arch.[24] Echocardiography is utilized as a confirmatory test, but cardiac catheterization and CT angiography are required for complete characterization of the AOPA [Figure 10]. Axial movie clip of the heart from a cardiac computed tomography angiogram in a 6-day-old male. The right pulmonary artery arises from the ascending aorta. The main pulmonary artery is separate from the aorta. The left pulmonary artery arises from the main pulmonary artery [Video 1].[24]
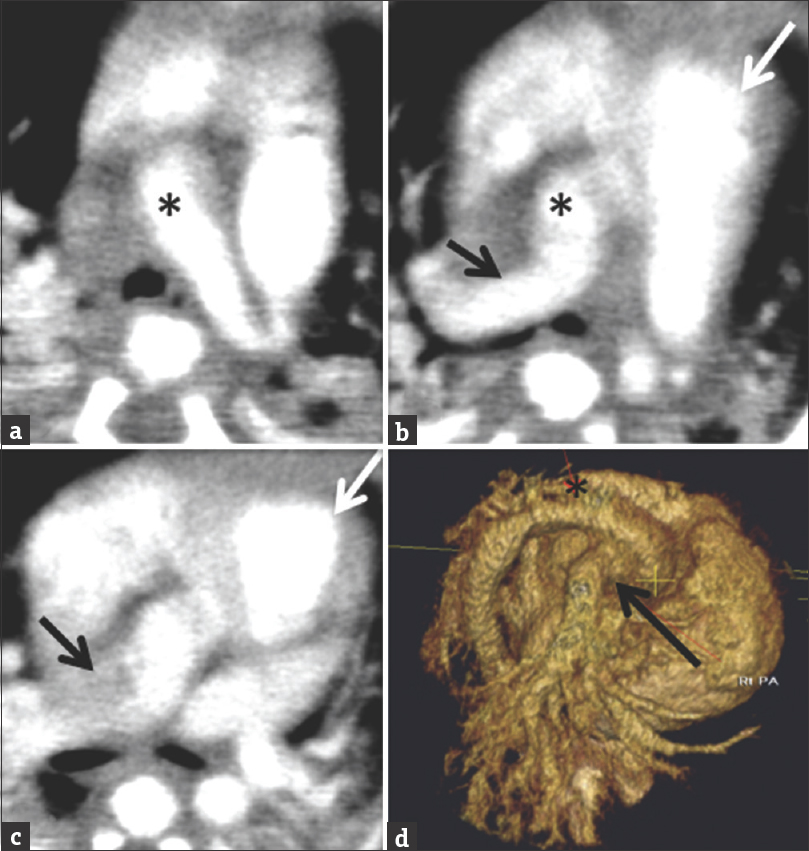
- Axial images and 3D reconstruction of the heart from a cardiac computed tomography angiogram in a 6-day-old male. The right pulmonary artery (black arrow) arising from the ascending aorta. The main pulmonary artery (white arrow) is separate from the aorta (black asterisk). The left pulmonary artery arises from the main pulmonary artery.
The prognosis for AOPA is poor if early surgical repair is not performed. Multiple surgical techniques exist and include direct reimplantation as well as creation of grafts or patches. A direct reimplantation of the anomalous branch PA to the MPA is preferred, if technically feasible.[24]
Left pulmonary artery sling
An LPAS, also known as an aberrant LPA, arises from the RPA, rather than from the MPA. The embryogenesis of an aberrant left pulmonary is thought to occur when the sixth aortic arch fails to form in utero. In addition to tracheobronchial anomalies, pulmonary slings have a 30% association with congenital heart defects.[25] Approximately 90% of patient with LPAS develop symptoms, and of those patients, the majority become symptomatic within the 1st year of life. Patients may present with signs of extrinsic airway obstruction, such as wheezing, dyspnea, and stridor.
Classification of LPAS is based on tracheobronchial anatomy. Type 1 has either normal tracheobronchial anatomy or a right prearterial or tracheal bronchus (type 1B) compressing both the right main bronchus and distal trachea leading to tracheomalacia with varying degrees of air trapping in the right lung, which is the “classic” pulmonary sling.[26]
Type 2 LPAS is more common than Type 1 LPAS. In type 2, there is a normal right upper lobe bronchus with a bridging bronchus arising from the left main bronchus that aerates the right middle and right lower lobes.[25] The aberrant LPA courses posterior to the bridging bronchus. Type 2 LPAS is further characterized by the presence of primary tracheal stenosis secondary to complete cartilaginous rings, a low tracheal bifurcation at the level of the T6 vertebra, and an inverted T-shaped carina.
Echocardiography is often the initial imaging investigation in LPAS, particularly in critically ill neonates due to its portability and high specificity. The origin of the aberrant LPA and its abnormal posterior course can be visualized on echocardiogram. A chest radiograph is also often performed early on given dyspnea at presentation. Type 1 cases may demonstrate fluid retention or air trapping in the right lung on radiography. Unilateral air-trapping is typically absent in Type 2. These patients may also present with dysphagia leading to the performance of a barium esophagram. The aberrant PA typically courses between the distal trachea and mid esophagus causing mass effect on the posterior wall of the trachea and anterior wall of the mid-esophagus which can be seen on fluoroscopy [Figure 11]. Cross-sectional imaging with CT or MR angiography adds value in surgical planning by depicting the anatomy of the chest and possible tracheobronchial anomalies [Figures 12 and 13].
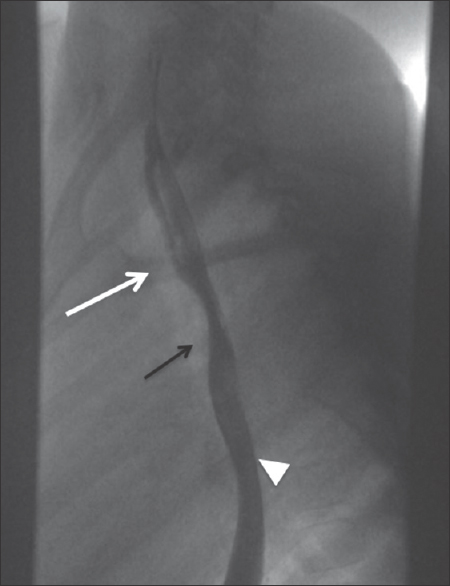
- Lateral esophagram on a patient with upper airway symptoms demonstrates an anterior impression, which can be seen with underlying anomalous left pulmonary artery (black arrow) between the airway (white arrow, outlined by an air column) and esophagus (white arrowhead).
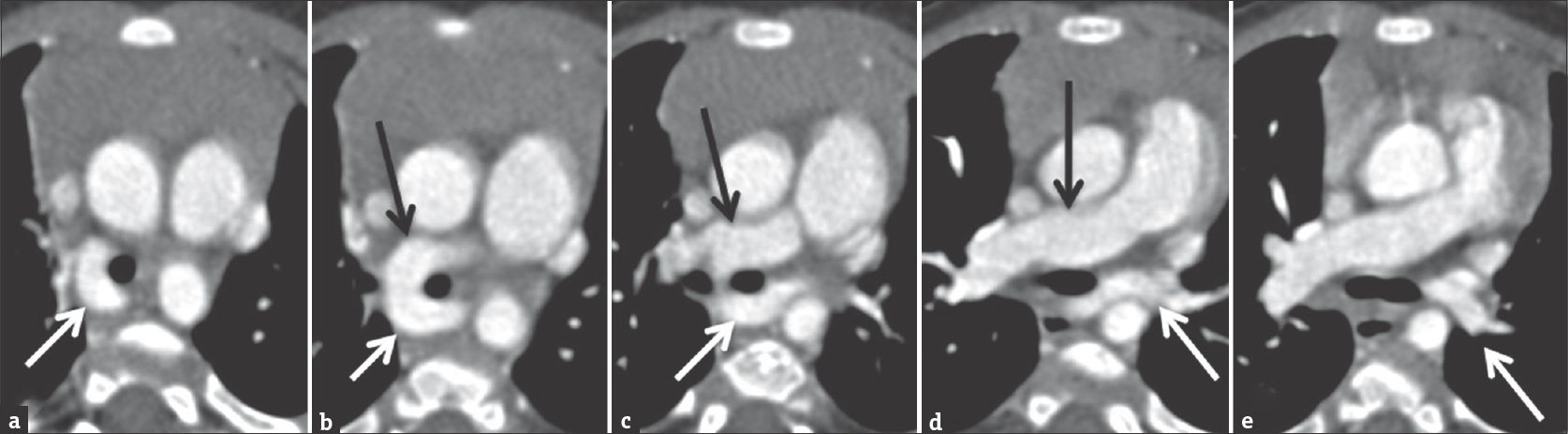
- Computed tomography angiogram of a 9-month-old male with prenatal concern for complex congenital heart disease and suspected left pulmonary sling on echocardiogram. Figures 12a to e are selected axial images demonstrating the anomalous origin of the left pulmonary artery (white arrow) from the right pulmonary artery (black arrow) as well as the abnormal posterior course of left pulmonary artery behind the trachea.
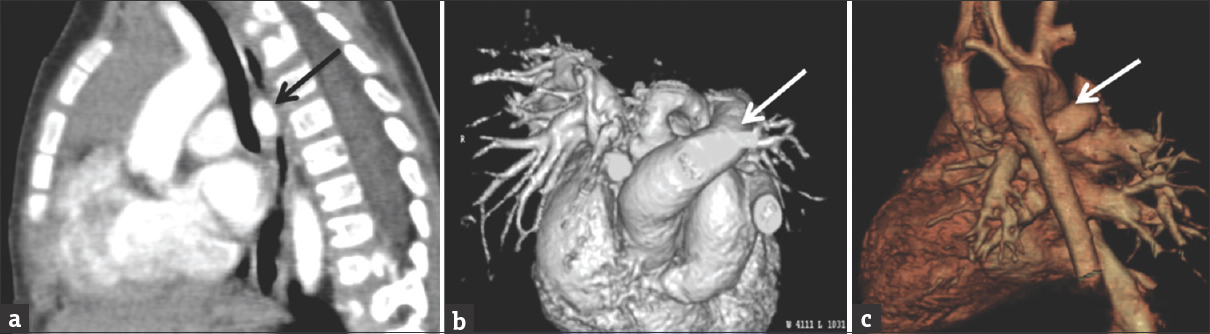
- Images from the same patient in Figure 13. Computed tomography angiogram shows the aberrant left pulmonary artery (black and white arrows) courses between the lower trachea and the mid-esophagus causing moderate tracheal stenosis. Figure 13b and c are 3D reconstructions of the computed tomography angiogram better demonstrating the pulmonary sling.
Surgical repair of LPAS involves reanastomosis of the left pulmonary to the MPA. Repair of the tracheal stenosis is performed at the time of the vascular repair for type 2 LPAS. Mortality in LPAS is determined by the need for tracheal repair. Patients not requiring tracheal surgery have an excellent prognosis as opposed to patients with long segment tracheal stenosis who have a mortality rate of 14%–22%.[26] Fortunately, as surgical techniques have advanced, including slide tracheoplasty, the mortality rate has dropped significantly. For example, Yong et al. reported a 0% operative and late mortality rate in at their institution from 2004 to 2011.[27]
CONCLUSION
We propose an approach to imaging of congenital PA anomalies [Table 3]. Initially, imaging evaluation should begin with chest X-ray and echocardiogram. Abnormalities identified on echocardiogram depending on the clinical scenario will require advanced imaging for surgical planning or optimized medical management.
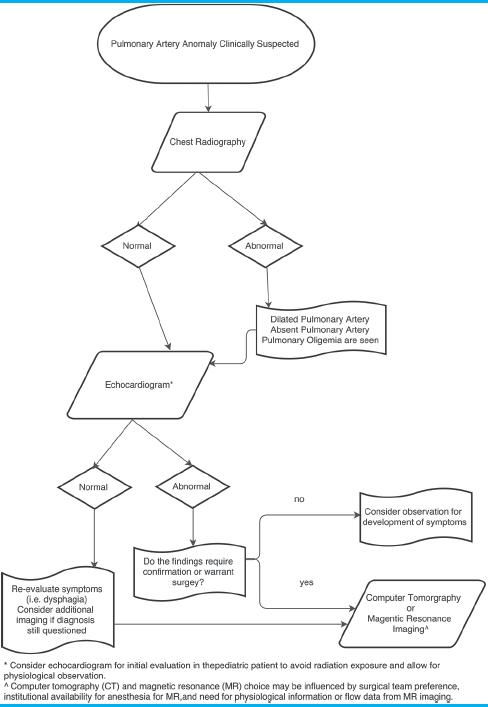
Congenital PA anomalies are an uncommon but important diagnosis for the pediatric and general radiologist to consider. These anomalies can occur in isolation or in association with congenital heart disease. While isolated anomalies may go unrecognized until adolescence or adulthood, those that occur in conjunction with congenital heart disease typically present earlier prompting an imaging evaluation.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Videos Available on: www.clinicalimagingscience.org
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2018/8/1/29/238184
REFERENCES
- High-quality low-dose cardiovascular computed tomography (CCT) in pediatric patients using a 64-slice scanner. Acta Radiol 2018:284185117752981. doi: 10.1177/0284185117752981
- [Google Scholar]
- Reference values for normal pulmonary artery dimensions by noncontrast cardiac computed tomography: The Framingham Heart Study. Circ Cardiovasc Imaging. 2012;5:147-54.
- [Google Scholar]
- Nomograms for two-dimensional echocardiography derived valvular and arterial dimensions in Caucasian children. J Cardiol. 2017;69:208-15.
- [Google Scholar]
- Cardiac valve disease: Spectrum of findings on cardiac 64-MDCT. AJR Am J Roentgenol. 2008;190:W294-303.
- [Google Scholar]
- Radiologic diagnosis of different types of pulmonary stenoses. Cardiovasc Radiol. 1977;1:45-57.
- [Google Scholar]
- 'Isolated’ pulmonary valve stenosis as part of more widespread cardiovascular disease. Br Heart J. 1976;38:472-82.
- [Google Scholar]
- Divided right ventricle: A review of its anatomical varieties. Pediatr Cardiol. 1984;5:197-204.
- [Google Scholar]
- Pathology of coronary arteries, myocardium, and great arteries in supravalvular aortic stenosis. Report of five cases with implications for surgical treatment. J Thorac Cardiovasc Surg. 1994;108:21-8.
- [Google Scholar]
- Percutaneous balloon valvuloplasty for pulmonic stenosis in adolescents and adults. N Engl J Med. 1996;335:21-5.
- [Google Scholar]
- Percutaneous transluminal balloon valvuloplasty for pulmonary valve stenosis. Circulation. 1984;69:554-60.
- [Google Scholar]
- Isolated right pulmonary artery discontinuity. Images Paediatr Cardiol. 2000;2:24-30.
- [Google Scholar]
- Congenital unilateral absence of a pulmonary artery. The importance of flow in pulmonary hypertension. Am J Cardiol. 1962;10:706-32.
- [Google Scholar]
- “Absent” pulmonary artery in one adult and five pediatric patients: Imaging, embryology, and therapeutic implications. AJR Am J Roentgenol. 2002;179:1253-60.
- [Google Scholar]
- Uncommon congenital and acquired aortic diseases: Role of multidetector CT angiography. Radiographics. 2010;30:79-98.
- [Google Scholar]
- Persistent diastolic flow reversal in abdominal aortic Doppler-flow profiles is associated with an increased risk of necrotizing enterocolitis in term infants with congenital heart disease. Pediatrics. 2007;119:330-5.
- [Google Scholar]
- Cardiovascular MR imaging of conotruncal anomalies. Radiographics. 2010;30:1069-94.
- [Google Scholar]
- Reinterventions after repair of common arterial trunk in neonates and young infants. J Am Coll Cardiol. 2000;35:1317-22.
- [Google Scholar]
- Idiopathic dilatation of the pulmonary artery – A case report. Echocardiography. 2013;30:E265-8.
- [Google Scholar]
- Anomalous origin of a pulmonary artery from the ascending aorta: Associated anomalies and pathogenesis. Am J Cardiol. 1988;61:850-6.
- [Google Scholar]
- The anomalous origin of the branch pulmonary artery from the ascending aorta. Interact Cardiovasc Thorac Surg. 2012;15:86-92.
- [Google Scholar]
- Pulmonary vascular sling: Report of seven cases and review of the literature. Pediatr Cardiol. 1989;10:81-9.
- [Google Scholar]
- Left pulmonary artery sling – Anatomy and imaging. Semin Ultrasound CT MR. 2010;31:158-70.
- [Google Scholar]
- Surgical management of pulmonary artery sling in children. J Thorac Cardiovasc Surg. 2013;145:1033-9.
- [Google Scholar]




