
Translate this page into:
Clinicoradiological Spectrum of L-2-Hydroxy Glutaric Aciduria: Typical and Atypical Findings in an Indian Cohort

Address for correspondence: Dr. Sniya Valsa Sudhakar, Department of Radiodiagnosis, Christian Medical College, Vellore - 632 004, Tamil Nadu, India. E-mail: sniya.sudhakar@gmail.com
-
Received: ,
Accepted: ,
Abstract
Context:
Neurometabolic disorders form an important group of potentially treatable diseases. It is important to recognize the clinical phenotype and characteristic imaging patterns to make an early diagnosis and initiate appropriate treatment. L-2-hydroxy glutaric aciduria (L2HGA) is a rare organic aciduria with a consistent and highly characteristic imaging pattern, which clinches the diagnosis in most cases.
Aims:
The study aims to describe the clinical profile, magnetic resonance imaging (MRI) patterns, and outcome in a cohort of children with L2HGA and to assess the clinicoradiological correlation.
Materials and Methods:
This is a retrospective descriptive study done at the Department of Radiodiagnosis and Neurological Sciences of our institution. Clinical and radiological findings of children diagnosed with L2HGA over an 8-year period (2010–2017) were collected and analyzed. Descriptive statistical analysis of clinical and imaging data was performed.
Results:
There were six girls and four boys. A total of 14 MRI brain studies in 10 patients with the diagnosis were analyzed. MRI of all patients showed a similar pattern with extensive confluent subcortical white-matter signal changes with symmetrical involvement of dentate nuclei and basal ganglia. In two children who presented with acute decompensation, there was asymmetric cortical involvement and restricted diffusion, which are previously unreported. There was no significant correlation between the radiological pattern with the disease duration, clinical features, or course of the disease.
Conclusion:
MRI findings in L2HGA are highly consistent and diagnostic, which helps in early diagnosis, particularly in resource-constraint settings, where detailed metabolic workup is not possible. The article also describes novel clinical radiological profile of acute encephalopathic clinical presentation.
Keywords
Dentate nucleus
L-2-hydroxy glutaric aciduria
leukodystrophy
magnetic resonance imaging
organic aciduria
subcortical white matter

INTRODUCTION
Organic acidurias form an important group of inherited metabolic disorders due to defect in intermediary metabolism of carbohydrate, amino acids, and fatty acids. Defective enzymes in specific pathways cause accumulation of organic acids in various tissues. The major clinical features are global developmental delay, encephalopathy, seizures, vomiting, failure to thrive, hepatomegaly, respiratory distress, and cardiac dysfunction. 2-hydroxy glutaric aciduria (2HGA) is a rare neurometabolic disorder which includes D2HGA, L2HGA, and combined D-L2HGAs.
L2HGA is a rare inherited neurometabolic disorder. Clinical course is usually chronic and gradually progressive with ataxia, spasticity, dystonia, seizures, macrocephaly, and intellectual disability. Magnetic resonance imaging (MRI) brain is diagnostic and often initiates further targeted genetic and metabolic workup. Hence, awareness of classic imaging findings is essential, in order to avoid diagnostic delay. In this article, we highlight the typical and atypical clinical presentations and imaging findings in an Indian cohort. We also describe an unusual clinical presentation of acute encephalopathy with previously unreported imaging findings, which further expands the clinicoradiological spectrum of L2HGA.
MATERIALS AND METHODS
A retrospective chart review of children diagnosed with L2HGA attending Pediatric Neurology clinic over a period of 8 years was done. The diagnosis was based on elevated L2HGA in urine which was performed based on the typical imaging findings. Various demographic details, clinical features, treatment details, and follow-up of the confirmed cases were documented. MRI brain with optimal T2, fluid-attenuated inversion recovery (FLAIR), and T1 sequences were available for all children. If patients had more than one examination, each was analyzed separately for temporal changes.
Fourteen MRI brain examinations from ten patients were available. A pediatric neuroradiologist with 6 years’ experience analyzed these images according to a predetermined reading template. White-matter abnormalities were assessed for pattern of distribution, lobar predilection, and sparing or involvement of structures such as corpus callosum, anterior limb of internal capsule (ALIC), and posterior limb of internal capsule (PLIC). Signal changes on T2, FLAIR, T1, and diffusion-weighted imaging were assessed. Cystic changes (FLAIR and T1 paralleling cerebrospinal fluid intensity) and rarefactions (hypointensity in FLAIR) were noted. Increased volume as evidenced by gyral broadening and volume loss as inferred from ventricular and subarachnoid space prominence were assessed.
Gray matter involvement was analyzed by location. Caudate, putamen, globus pallidus, thalamus, dentate nucleus, and cortex were separately assessed. Brainstem and cerebellar changes were also assessed. Global cerebellar (vermian and hemispheric) volume loss was documented. MR spectroscopy (MRS), postcontrast studies, and spine imaging were analyzed whenever available.
RESULTS
Ten children were included. Diagnosis was confirmed by urine gas chromatography-mass spectrometry (GC-MS) and characteristic imaging findings. In one child, diagnosis was further confirmed by genetic testing.
Demographic and clinical characteristics
There were six girls and four boys. Mean age at the onset of symptom was 2.1 years (7 months–5 years). Mean age at diagnosis was 8 years (1–10 years); mean duration of delay in diagnosis was 5.5 years (0–11 years). Misdiagnosis at initial presentation as mitochondrial cytopathy, Canavan disease, and Alexander’s disease was common. Clinical features and course of the disease are tabulated [Table 1]. The most common initial presentation was recurrent febrile seizures in five (50%) children followed by progressive gait ataxia in two (20%), intellectual disability in two (20%), and acute encephalopathy in one (10%). There was a mild motor developmental delay in all (100%) children; none of them had adverse perinatal events. Family history was positive in four children (Cases 2, 8 and 4, 5 are siblings). The percentages of clinical signs and symptoms are depicted in the bar diagram [Graph 1].
| Patients | Sex | Age at the onset of symptoms | Presenting symptoms | Macrocephaly | Spasticity | Ataxia | Seizures | Extrapyramidal involvement | Intellectual disability | Acute decompensation | Follow-up duration | Treatment | Treatment response |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Case 1 | Female | 1 year | Febrile status epilepticus | + | - | + | Febrile status epilepticus | - | - | - | 5 months | + | Improving |
| Case 2 | Male | 2 years | Acute encephalopathy | + | + | + | Right focal seizures (febrile + afebrile) | + | + | + | Expired | - | Expired |
| Case 3 | Female | 4 years | Progressive ataxia | - | + | + | Febrile seizures | - | - | - | 7 years | + | Stable |
| Case 4 | Male | 7 months | Febrile seizures | + | + | + | Febrile seizures | - | + | - | 7 years | + | Stable |
| Case 5 | Male | 2 years | Intellectual disability | + | + | + | - | - | + | - | 7 years | + | Stable |
| Case 6 | Female | 7 months | Febrile seizures | - | - | + | Febrile and afebrile seizures | - | - | - | 8 months | + | Improving |
| Case 7 | Male | 11 months | Febrile seizures | + | + | + | Febrile seizures (generalized + right focal) | - | + | + | 7 years | + | Worsening |
| Case 8 | Female | 5 years | Intellectual disability | - | + | + | - | - | + | - | 8 years | + | Stable |
| Case 9 | Female | 3 years | Progressive ataxia | + | + | + | Afebrile seizures | + | + | - | 5 months | + | Stable |
| Case 10 | Female | 2 years | Febrile seizures | + | + | + | Febrile seizures | - | - | - | 2 years | + | Stable |
-: Absent, +: Present
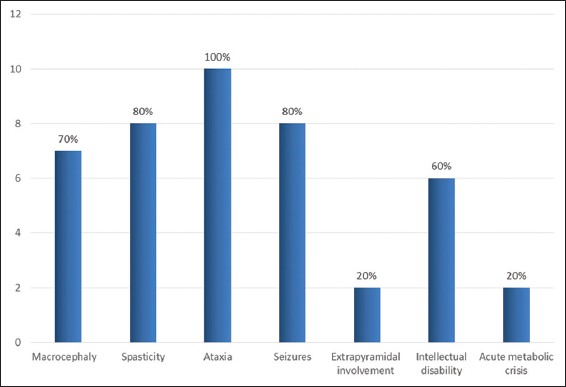
- Clinical symptoms and signs in children with L-2-hydroxy glutaric aciduria.
Acute metabolic crisis was seen in two (20%) children, one of whom succumbed to the illness. One child presented initially with acute encephalopathy and the other developed encephalopathy during follow-up. Description of the cases with acute encephalopathic presentation is detailed later.
Magnetic resonance pattern imaging pattern
A total of 14 MRIs were available for analysis (four out of ten children had two MRs). The mean interval between the MRI of these four children was 3.75 years (range of 2–5 years). Mean age at MRI was 8.4 years (range of 9 months–15 years). Mean disease duration at MRI was 6.23 years (1 month–14 years).
White-matter involvement
Subcortical white matter was involved in all cases (100%), followed by deep white matter in 92.8%, while periventricular white matter was spared in all except two MRs (14.2%) The subcortical involvement was seen in all lobes except in one MRI where occipital lobe was spared. White-matter involvement was most confluent in the frontal lobes followed by parietotemporal lobes, whereas occipital involvement was least confluent, paralleling the severity of lobar involvement.
Rarefaction and cystic changes were common in our series with 92.8% and 78.6%, respectively. Rarefaction was frontal predominant in most cases, while cystic changes were sometimes more pronounced in temporal and parietal lobes [Figure 1]. Gyral broadening related to white-matter swelling was seen in 50% of MRs again with frontal predilection. One child showed asymmetric left-sided white-matter swelling involving frontal temporal and parietal lobes both in the acute phase and in the previous MRI done 5 years before.
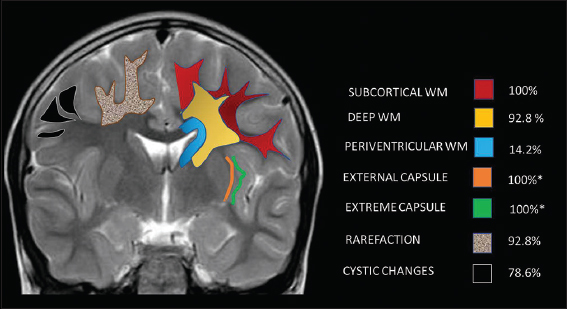
- Pictorial illustration of distribution and pattern of white-matter involvement.*12/12 children above 1 year.
There was symmetric involvement (78.6%) in all, except three MRIs, two of which were during acute presentation and the third was the initial MRI of one child (case 7) who later presented with acute decompensation. External and extreme capsules were involved in all MRIs done beyond infancy. MRIs of two children done at 8 months and 1 year of age were not assessed for this finding due to ongoing myelination.
Along with periventricular white matter, other deeply located white-matter structures were mostly spared. Corpus callosum and PLIC showed signal changes in one during the acute phase, while callosal thinning was apparent in 42.8%. ALIC was however commonly involved (78.6%). Anterior commissure, cerebellar, and brainstem white matter were never involved [Table 2, Figure 2].
| MRI findings | Age at MR | Duration of disease at MR | Subcortical white matter | Deep white matter | PVWM | Confluence | Rarefaction | Cystic changes | Symmetry | Swelling | Corpus callosum | External/extreme capsule | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F | P | T | O | F | P | T | O | F | P | T | O | F | P | T | O | Lobe | Lobe | Lobe | ||||||
| Case 1 | 1 year | 1 month | + | + | + | + | + | + | + | + | - | - | - | - | + | + | - | - | FPT | FPT | + | F | - | NA |
| Case 2 | 9 years | 7 years | + | + | + | + | + | + | + | + | +L | +L | +L | +L | + | + | +L | +L | - | FP | -L | F | +* | + |
| Case 3 | 7 years | 3 years | + | + | + | + | + | + | + | + | - | - | - | - | + | - | - | - | FPT | TFP | + | - | +Thin | + |
| 12 years | 8 years | + | + | + | + | + | + | + | + | - | - | - | - | + | - | - | - | FPT | TFP | + | - | +Thin | + | |
| Case 4 | 13 years | 12 years | + | + | + | + | + | + | + | + | - | - | - | - | + | + | - | - | FPT | FPT | + | - | +Thin | + |
| 15 years | 14 years | + | + | + | + | + | + | + | + | - | - | - | - | + | + | - | - | FPT | FPT | + | - | +Thin | + | |
| Case 5 | 13 years | 11 years | + | + | + | - | + | - | - | - | - | - | - | - | + | + | + | - | FPT | - | + | - | - | + |
| Case 6 | 9 months | 2 months | + | + | + | + | - | - | - | - | - | - | - | - | + | + | + | + | FPT | - | + | F | - | NA |
| Case 7 | 6 years | 5 years | + | + | + | + | + | + | + | + | - | - | - | - | + | + | + | + | FPT | FTP | -L | FTPL | - | + |
| 11 years | 10 years | + | + | + | + | + | + | + | + | - | +L | +L | +L | + | + | + | + | PTOR | PTOR | -L | FTPL | - | + | |
| Case 8 | 8 years | 3 years | + | + | + | + | + | + | - | - | - | - | - | - | + | + | + | - | FPT | PTF | + | - | - | + |
| Case 9 | 7 years | 4 years | + | + | + | + | + | + | + | + | - | - | - | - | + | + | + | - | FTP | PTF | + | F | +Thin | + |
| 10 years | 7 years | + | + | + | + | + | + | + | + | - | - | - | - | + | + | + | - | FTP | PTF | + | F | +Thin | + | |
| Case 10 | 3 years | 1 year | + | + | + | + | + | - | - | - | - | - | - | - | + | + | + | - | FPT | - | + | - | - | + |
PVWM: Periventricular white matter
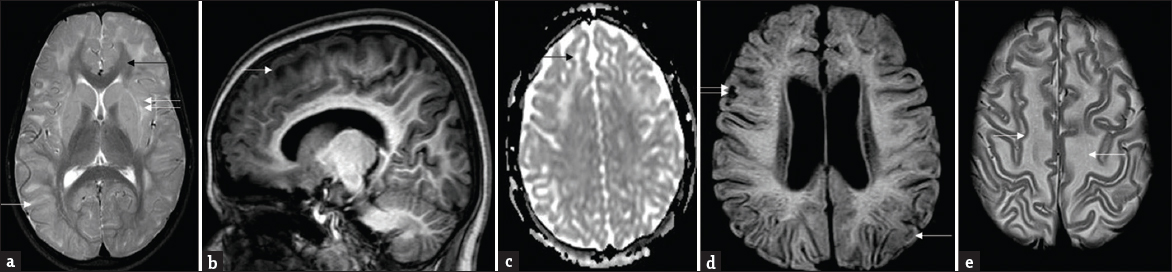
- Patterns of white-matter involvement: Composite representative images from our series: T2 axial (a) shows symmetrical subcortical white-matter hyperintensities (white arrow) sparing deep and periventricular white matter, posterior limb of internal capsule, and corpus callosum (black arrow). Note signal changes of external and extreme capsule (double arrows). T1 sagittal image (b) shows subcortical and frontal predominance. Apparent diffusion coefficient map (c) shows facilitated diffusion (black arrow). Note small areas of rarefaction (white arrow) and cystic changes (double arrows) involving subcortical white matter on fluid-attenuated inversion recovery (d). Also seen is relative prominence of lateral ventricles. T2 axial images at supraventricular level (e) demonstrates mild swelling of frontal white matter (white arrows).
Gray matter involvement
Dentate nucleus was involved in all cases (100%). The dentate hilus was spared in all except one, this case showed extensive involvement of the rest of the white matter. The caudate and putamen were invariably involved. One case showed central predominant striatal involvement. Globus pallidus (GP) was involved in all cases beyond 1 year of age. Nearly 61.5% showed sparing of the GP lamina. Thalamus was involved in one case (7.1%) along the paramedian aspect. Cerebral cortical involvement was seen in two (14.3%) cases during acute presentation. The spectrum of gray matter involvement is depicted in Graph 2 and Figure 3.
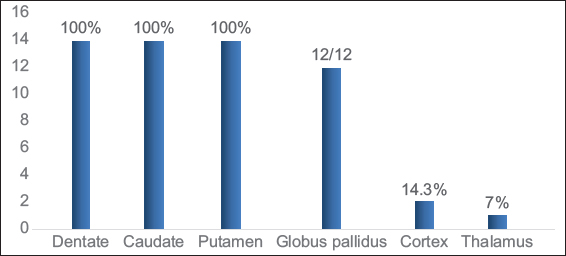
- Gray matter involvement in L-2-hydroxy glutaric aciduria.
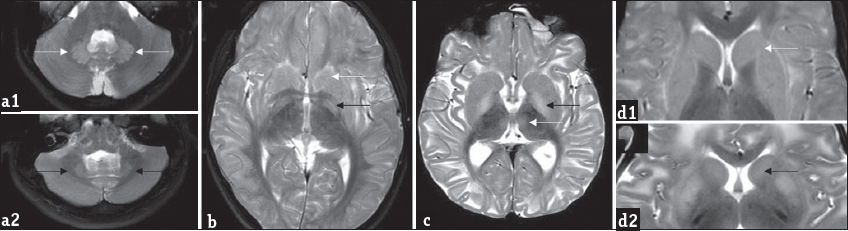
- Patterns of gray matter involvement: Composite representative images from our series: Bilateral symmetric dentate hyperintensity is seen on T2 axial images in two patients (a1 and a2), while mild swelling is appreciable in a1, relative volume loss is seen in a2. T2 axial image (b) shows bilateral caudate, putamen, and globus pallidus involvement (white arrow). Note the laminar sparing of globus pallidus (black arrow). T2 axial image (c) in another patient shows absence of laminar sparing of globus pallidus (black arrow) as well as bi-thalamic paramedian signal changes (white arrow). T2 axial (d1) images show peripheral rim of hyperintensity along the caudate and putamen (white arrow), T2 axial (d2) images in another patient show central predominant involvement (black arrow).
Other features
Global cerebellar volume loss was seen in 42.8% of cases.
Diffusion study was available in 64.3% of cases. Diffusion restriction was seen in four cases, two (cases 2, 7) of whom presented with acute decompensation. White matter-restricted diffusion was seen in both cases; one revealed additional globus pallidus involvement. The third child showed restricted diffusion of the caudate, putamen, and hippocampi in an MRI done immediately after an ictus. Isolated globus pallidus-restricted diffusion was noted in the fourth child [Figure 4].
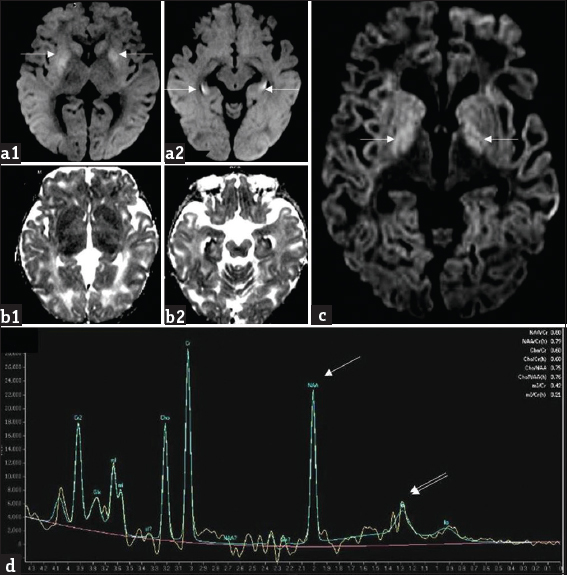
- A 1-year-old girl with L-2-hydroxy glutaric aciduria presenting with febrile status epilepticus: Diffusion-weighted imaging (a1, a2) and apparent diffusion coefficient (b1, b2) images show symmetric bilateral restricted diffusion involving caudate and putamen as well as along the periphery of hippocampi (white arrows). Axial diffusion-weighted imaging image (c) from another patient, a 10-year-old girl with progressive ataxia shows milder signal changes of globus pallidus (white arrows). Magnetic resonance spectroscopy (d) from the same patient shows reduction in NAA peak at 2 ppm (white arrow) and presence of lipid lactate peak at 0.8–1.4 ppm (double arrows).
Postcontrast study was available in eight cases (57%) and none showed significant parenchymal or meningeal enhancement. Gradient sequences (available in 57%) showed no signal changes.
MRS showed mild N-acetylaspartate (NAA) reduction and small lipid-lactate peak in two cases and was unremarkable in others [Figure 4]. Spine imaging available in two cases was unremarkable.
None of the cases in this series showed coexisting brain tumor. One case had incidental left periventricular nodular heterotopias, the largest measuring 6 mm [Figure 5]. Four computed tomography (CT) studies were available from two patients. Frontal and subcortical hypodensity was apparent in both the CTs of one child. The other two CTs were from the child who presented with acute decompensation, details of which are discussed below.
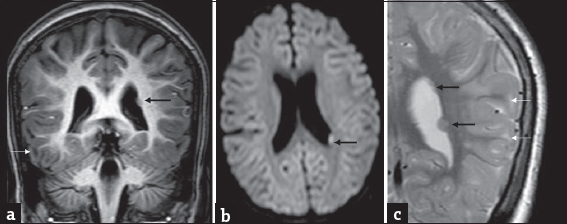
- A 7-year-old girl with progressive ataxia and past history of febrile seizures diagnosed with L-2-hydroxy glutaric aciduria : Incidental finding of left subependymal nodular heterotopia on magnetic resonance imaging. T1 coronal (a) axial diffusion-weighted imaging (b) and T2 coronal (c) show small nodular subependymal heterotopia (black arrows) paralleling gray matter signal. T2 image also shows smaller heterotopias more superiorly. Note the subcortical white matter signal changes related to L-2-hydroxy glutaric aciduria (white arrows) seen in all images.
In our series, digression from typically described imaging features was observed in cases with acute presentation. Asymmetry of white-matter involvement with diffusion restriction and involvement of cortex and deeper white-matter structures were found to be significantly associated with acute presentation. The clinicoradiological profile of children with acute encephalopathic presentation (cases 2 and 7) is detailed below.
Case 2
A 9-year-old male child presented with recurrent right focal motor seizures followed by right hemiparesis and altered sensorium, which was precipitated by a febrile illness. Historically, he had a mild developmental delay and nonprogressive ataxia since 4 years of age. Evaluation revealed acidosis and elevated liver enzymes (serum glutamic pyruvic transaminase 3061 U/L and serum glutamic-oxaloacetic transaminase 3859 U/L). Evaluation for alternative etiologies was noncontributory. CT and MRI findings are described in Figure 6. His sensorium and liver functions gradually improved over 2 weeks with supportive treatment. Basic metabolic workup was within normal limits. He was not initiated on any definitive treatment as the etiology was undetermined. Two months later, he again developed recurrence of right focal seizures followed by prolonged encephalopathy with acidosis and succumbed to the illness. Repeat CT brain findings are depicted in Figure 6. Liver functions were normal during this admission. Case 8 is his younger sibling, who presented at 8 years of age with poor scholastics who was evaluated further and confirmed with L2HGA, initiated on treatment, and is currently stable at 8-year follow-up.
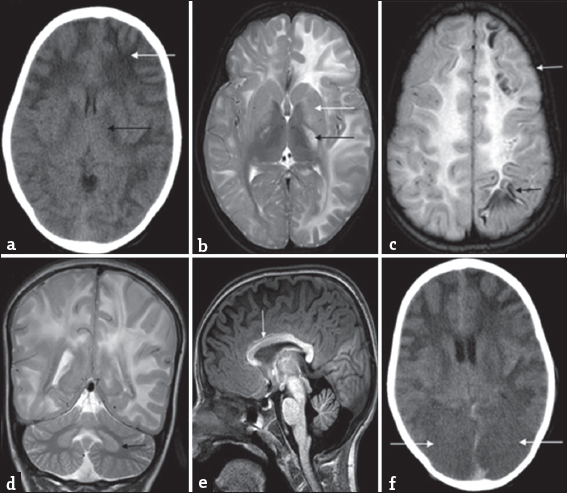
- Serial imaging findings in a 9-year-old boy with acute encephalopathy and seizures (Case 2): Initial computed tomography (a) shows extensive asymmetric white-matter involvement with left-sided predominance (white arrow). Posterior limb of internal capsule involvement (black arrow) is also appreciable along with lateral ventricular effacement. Magnetic resonance imaging brain done 1 month later (b-e) again shows extensive asymmetric white-matter involvement with left-sided predominance. Note the periventricular involvement, confluent nature, and swelling appreciable on the left side. Bilateral asymmetric posterior limb of internal capsule involvement is also noted (black arrow) along with signal changes in basal ganglia (white arrow) and thalamic sparing on T2 axial image (b). Fluid-attenuated inversion recovery image (c) shows subcortical cystic changes in frontal and parietal lobes (black arrow). Also note the cortical swelling and signal changes in the left frontal lobe (white arrow). T2 coronal image (d) shows bilateral dentate involvement (black arrow). T1 sagittal image (e) shows anterior predominant callosal involvement (white arrow) as well as frontal predominance of white-matter involvement. Subsequent computed tomography done 2 weeks later (f) shows bilateral occipital hypodensities with loss of gray-white differentiation (white arrows), possibly representing posterior cerebral artery infarcts secondary to transtentorial herniation.
Case 7
An 8-year-old male child presented with recurrent febrile seizures since 11 months of age and progressive ataxia from 3 years of age. Along with classic features, his initial MRI showed asymmetric distribution as shown in Figure 7. Diagnosis was further confirmed by highly elevated L2HG in urine. He was initiated on carnitine, riboflavin, and low-protein diet. He remained stable till 13 years of age and presented with acute onset of fever precipitated right focal seizures with secondary generalization. He became encephalopathic with acidosis requiring hospitalization and supportive management. Although his sensorium improved gradually over 4 days, he had worsening of ataxia and spasticity along with right hemiparesis. Repeat imaging revealed new findings as shown in Figure 7. Riboflavin and carnitine doses were further increased without much clinical response and he became bedbound at 6-month follow-up.
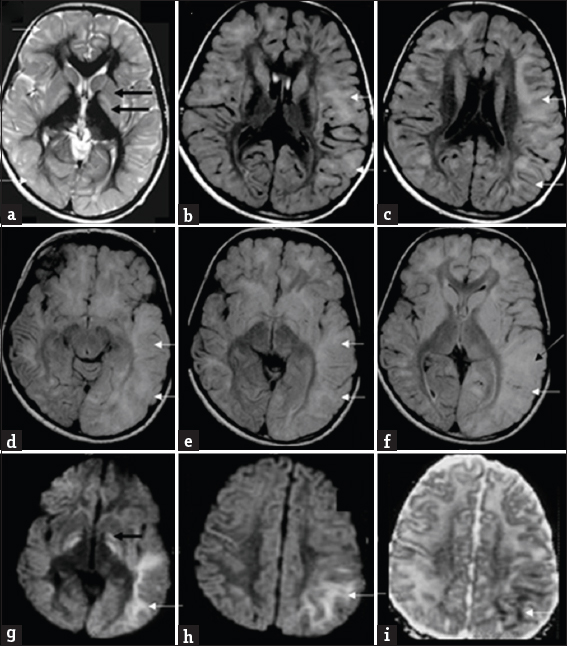
- Magnetic resonance imaging findings in an 8-year-old boy presenting with progressive ataxia (Case 7): Initial magnetic resonance imaging shows subcortical predominant white-matter involvement (white arrows) along with symmetric signal changes of caudate, putamen, and globus pallidus (black arrows) on T2 axial images (a). Axial fluid-attenuated inversion recovery images (b and c) show asymmetry of white matter changes, with left side showing more confluent and extensive findings in posterior frontal and parieto-temporal regions Axial fluid-attenuated inversion recovery images (d-f) from subsequent magnetic resonance imaging after 5 years during the acute presentation shows extensive asymmetric gyral swelling and confluent white-matter signal changes on the left side with parietotemporal predominance (white arrows). Also note the signal changes of cortex (white arrow) in f. Diffusion-weighted imaging (g and h) and apparent diffusion coefficient (i) image also show restricted diffusion within the white matter (white arrows) as well as bilateral globus pallidus (black arrow).
Results of comparative analysis of MRI features between acute and nonacute groups are tabulated [Table 3].
| Imaging findings | Acute (n=2), n (%) | Nonacute (n=8), n (%) |
|---|---|---|
| Symmetry of white-matter involvement | Asymmetric 2/2 (100) | Asymmetric (1/12) 8.3 |
| Cortical involvement | Present 2/2 (100) | Present 0/12 (0) |
| Restricted diffusion | Present 2/2 (100) | Present 2/12 (16.6) |
| Involvement of deeper white matter structures (corpus callosum, PVWM, PLIC) | Present 2/2 (100) | Present 0/12 (0) |
PVWM: Periventricular white matter, PLIC: Posterior limb of internal capsule, MR: Magnetic resonance
Clinicoradiological correlation
Statistical analysis was not done in view of small sample size; however, there is no apparent correlation between disease duration, seizures, or clinical course with MRI findings such as extent of white-matter involvement, rarefaction, and cystic changes. Four children had serial MRIs with a mean interval of 3.75 years (range of 2–5 years). No significant interval changes were appreciable in three of them, while the fourth child (Case 7) showed interval progression of extent of deep white-matter involvement bilaterally along with acute decompensation-related changes [Figure 8].
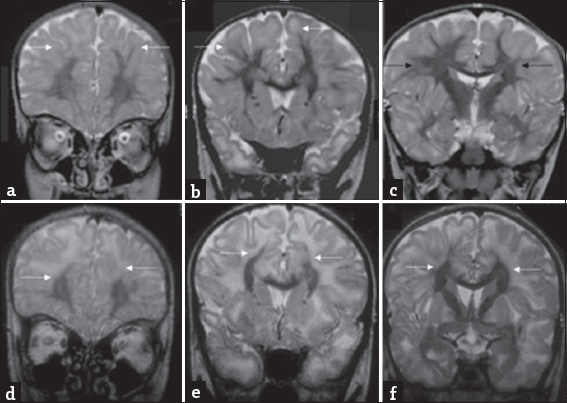
- An 8-year-old boy presenting with progressive ataxia: Temporal evolution of white matter changes over 5-year interval. Initial magnetic resonance imaging shows confluent subcortical predominant white-matter hyperintensity (white arrows) with relative sparing of deep white matter (black arrows) on T2 coronal images (a-c). T2 coronal images at the same levels (d-f) from subsequent magnetic resonance imaging done after 5 years show more extensive deep white matter involvement (white arrows).
Follow-up and treatment response
All children except the one who expired during acute crisis were initiated on carnitine (50–100 mg/kg/day) and riboflavin (100–200 mg/day). The mean duration of follow-up was 4.3 years (5 months–8 years). Six (60%) children remained stable with treatment, two (20%) had significant improvement, and one (10%) had worsening of symptoms.
DISCUSSION
L2HGA is a rare organic aciduria with a gradually progressive clinical course and characteristic imaging findings. Duran et al., in 1980 reported the case of a 5-year-old boy with nonspecific mental and motor retardation with persistent high urinary excretion of L2HG.[1] Barth et al., in 1992 described a cohort of children with similar phenotype and biochemical and classic imaging pattern.[2] Since then, there have been several case reports and series, worldwide, of children and adults with L2HGA. The largest cohort reported till date is by Steenweg et al.[3,4]
Metabolism, gene, and pathogenesis
The biochemical pathway was first postulated by Rzem et al., that L2HG could be formed from 2 ketoglutarate (2KG).[5] The findings were further confirmed by the same group.[6] It was hypothesized to be an unwanted side reaction of L-malate dehydrogenase in tricarboxylic acid cycle, repaired by L2 hydroxy glutarate dehydrogenase. With no known physiological functions, L2HGA is regarded as a “disorder of metabolite repair”[Figure 9]. Moderate elevation of lysine probably reflects low mitochondrial 2-KG availability.[7] The gene involved is L2HGDH gene (C14orf160), located on chromosome 14.[8-10] Around seventy disease-causing variants have been described.[4] The exact pathogenesis of leukodystrophy is poorly understood, though oxidative stress might play an important role.[11]
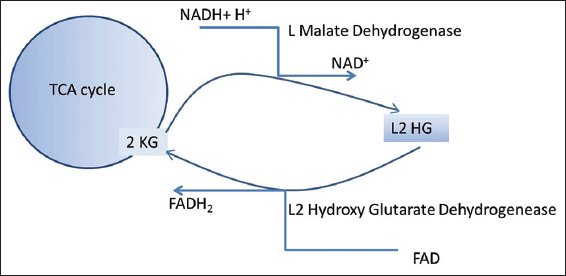
- Metabolic pathway of L-2-hydroxy glutaric aciduria.
Clinical features
The cardinal clinical manifestations are developmental delay, seizures, slowly progressive ataxia, spasticity, intellectual disability, and extrapyramidal features. The demographic and clinical features in our series are concordant with that of previous literature.[3] Macrocephaly though not obligatory is a common association.
Atypical presentations such as dystonia, anxiety, autism, status epilepticus, and acute hemiplegia hemi convulsion epilepsy syndrome (HHE) have been reported.[12-16] One child in our series presented with febrile status epilepticus. While the largest case series does not describe an acute presentation, there are several case reports of L2HGA with severe neonatal encephalopathy, sudden death in infancy, encephalopathy with rapid functional decline following seizures, and trauma.[17-20] Our series had two children (20%) with acute presentation and documented metabolic acidosis, which was not described in any of the aforementioned cases. Both the children had features of acute HHE syndrome, though they did not improve with time as reported by Lee et al.;[16] while one succumbed to the illness, the other had a rapid functional decline in our series. We would like to highlight this as a possible presenting symptom like any other organic acidurias. Such a presentation calls for change in the management protocol to include hydration, cessation of protein intake, gut sterilization measures, dialysis, and more importantly precautionary measures to follow during intercurrent illnesses.
Imaging findings
Imaging findings of L2HGA are usually consistent and diagnostic. Frontal predominant subcortical white-matter hyperintensities with sparing of periventricular, commissural, brainstem, and cerebellar white matter are characteristically seen. The involved white matter shows variable degrees of swelling, rarefaction, cystic changes, and volume loss. Dentate and basal ganglia are involved in almost all patients, while thalamus is typically spared. Cerebellar volume loss is often associated. The main imaging features of the cases with chronic clinical course in our series are consistent with that of previous literature.[3,21,22]
While acute encephalopathy has been previously reported, imaging features during acute presentation have not been documented. Significant asymmetry, cortical involvement, involvement of deep white-matter structures such as PLIC and corpus callosum, and restricted diffusion seen in our series appear to be consistently associated with acute clinical presentation. While the exact pathogenesis remains unknown, the asymmetry observed on imaging could explain HHE presentation of our cases.
Another finding which is not alluded to in detail previously is the involvement of ALIC. Normal myelinated ALIC was not appreciable in most of our cases. This could have contributed to the peripheral rim sign described along the striatum in previous reports.[4,21]
MRS findings in our series were limited to NAA reduction and a peak at 0.8–1.4 ppm in two cases. The relative contributions of lipid and lactate in the latter peak could not be inferred due to lack of optimal intermediate TE images. NAA reduction has been attributed to axonal loss secondary to spongiosis.[23,24] Lipid peak can be present during active demyelination and lactate elevation has been described. Glutamate/L2HG peaks at 2.1–2.5 ppm, alterations in Myo-inositol (MI), and choline peaks as described in literature were not apparent in our series.
Facilitated diffusion was seen in most of the involved white matter as described previously. Restricted diffusion was seen in three MRIs in our series unlike the previous reports. One prior report describes globus pallidus restriction which was attributed to myelin splitting and vacuolation in contrast to demyelination with cystic cavitation seen in white matter.[23] However, later articles allude to the absence of significant differences between pathological processes involving the white matter and striatum except in severity.[24] The basis of striatal restricted diffusion seen in three of our cases remains unknown. Cases 2 and 7 who presented with acute encephalopathy also showed extensive unilateral diffusion restriction in parieto-occipito-temporal white matter, likely related to cytotoxic edema due to rapid accumulation of toxic metabolites, though the asymmetry of cerebral involvement is intriguing and remains unexplained.
Tumor and autopsy findings
L2HGA has a possible role in tumorigenesis with a reported tumor prevalence of 5%.[25,26] Astrocytoma, oligodendroglioma, ependymoma, medulloblastoma, and even non central nervous system malignancy such as Wilms tumor are known associations.[26-28] There are autopsy reports revealing spongiosis and cystic cavitations in subcortical white matter, with minimal abnormalities of the basal ganglia and dentate nucleus without abnormal storage in neurons and glial cells.[20,29,30] Neonatal autopsy had also revealed gliosis in the pontine and inferior olivary nuclei.[17]
Correlation
Although the clinical phenotype appears homogenous, correlation between phenotype, biochemical parameters, radiological and genotypic features in this disorder has been difficult to establish with the available literature.[4,31] Moroni et al., had observed a good correlation between disease severity and extent of MRI lesion, though the same has not been found in other studies.[32] Statistical correlation analysis was deferred in our series due to limited sample size.
Treatment response
There have been reports of significant clinical response with riboflavin.[12,13,33,34] Treatment response might be influenced by the residual enzyme activity based on the type of mutation, though a consistent correlation has not been established. Genetic workup done in one child in our series revealed a homozygous missense variation in exon 10 of L2HGDH gene; this child is stable on treatment with no seizure recurrence and has developmental gains at 6-month follow-up. It is prudent to give riboflavin and carnitine trial in all patients based on the anecdotal reports.
D2 hydroxy glutaric aciduria
D2HGA, another form of 2HGA, presents clinically with developmental delay, hypotonia, seizures, and cardiomyopathy. Urine GC-MS reveals elevated levels of 2HGA, and further chiral confirmation needs to be done to differentiate D2 and L2 enantiomers. Although the clinical presentation often can suggest either D2-HGA or L2-HGA, MRI helps in further delineating the disorder. MRI brain findings in D2-HGA include enlargement of the lateral ventricles, subdural effusions, subependymal pseudocysts, signs of delayed cerebral maturation, cerebral white-matter abnormalities, and callosal agenesis.[10]
Differential diagnosis
Other inherited metabolic disorders can mimic L2GHA clinically and radiologically.[3,35] The main imaging differentials include Canavan disease, Kearns–Sayre syndrome (KSS), and 3-hydroxy-3-methylglutaryl-coenzyme (HMGCoA) lyase deficiency. Although the clinical presentations are different, involvement of subcortical white matter is seen in all. In Canavan disease, white-matter involvement is diffuse and confluent; globus pallidus and thalamus are involved, and caudate, putamen, and dentate nucleus are spared. In KSS, subcortical white-matter hyperintensities are seen along with prominent globus pallidus, caudate, thalamus, dentate, and brain stem involvement. In HMGCo A lyase deficiency, white-matter hyperintensity is seen along with caudate and putamen, whereas brain stem and cerebellum are spared. Detailed descriptions of these disorders are compared in Table 4 and Figure 10.
| L2HGA | Canavan disease | Kearns-Sayre syndrome | HMGCo A lyase deficiency | |
|---|---|---|---|---|
| Clinical features | Manifests in early childhood with macrocephaly, slowly progressive ataxia, spasticity, dystonia and seizures | Presents in early infancy with macrocephaly, loss of acquired milestones, hypotonia, visual impairment, and decerebrate posturing | Symptom onset before 20 years of age with progressive external ophthalmoplegia, deafness, ataxia, and cardiac conduction defects | Manifests in early infancy with recurrent episodes of lethargy with severe acidosis, hypoglycemia, and elevated liver enzymes |
| White-matter involvement | Subcortical and frontal predominance Periventricular, callosal and PLIC sparing |
Diffuse confluent white-matter involvement Subcortical predominance can be present No frontal predilection |
Subcortical predominance | Frontal and periatrial predominant |
| Gray-matter Involvement | Caudate, putamen, globus pallidus, and dentate involvement Thalamic sparing |
Globus pallidus and thalamus involvement Caudate, putamen, and dentate sparing |
Globus pallidus, caudate, thalamus usually involved Less prominent putaminal involvement |
Dentate and caudate involvement |
| Brain stem and cerebellar white matter | Spared invariably | Invariably involved | Usually involved | Usually spared |
L2HGA: L-2-hydroxy glutaric aciduria, PLIC: Posterior limb of internal capsule
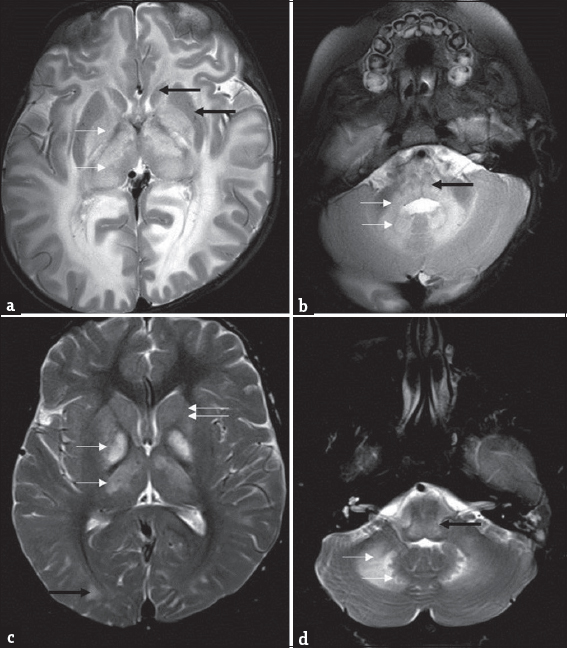
- Differential diagnosis of L-2-hydroxy glutaric aciduria. Imaging features of an 1-year-old boy with macrocephaly and neuroregression diagnosed as Canavan disease; a 9-year-old boy with progressive external ophthalmoplegia, cardiac conduction defects, and deafness diagnosed as Kearns–Sayre syndrome: T2 axial images of Canavan disease (a and b) show extensive panlobar white-matter hyperintensity in subcortical, deep, and periventricular distribution along with symmetric globus pallidus and thalamic hyperintensity and swelling (white arrow). Caudate and putamen are spared (black arrows). Significant medullary (black arrow), cerebellar white matter (white arrows), and dentate hilus signal changes are noted. T2 axial images of Kearns–Sayre syndrome at the same levels (c and d) show symmetric globus pallidus and thalamic hyperintensity (white arrows), while caudate and putamen are relatively spared (double arrows). Note the subcortical predominant white-matter involvement in the occipital regions (black arrow). Medulla (black arrow), cerebellar white matter, and dentate nucleus (white arrows) also show hyperintense signal changes.
Limitations
Relatively small sample size and retrospective nature were the main limitations. Although MRI studies were not homogeneous in protocol, availability of optimal primary sequences (T2, FLAIR, and T1 images) and systematic analysis allowed reasonable comparison. Absence of histopathology is another drawback, particularly in cases with acute decompensation. Availability of genetic data would have enhanced the value of the study allowing correlation with outcome.
CONCLUSION
This article reemphasizes the role of MRI in early diagnosis and presents novel clinical and radiological features of acute encephalopathic presentation in L2HGA. Early diagnosis and treatment initiation could potentially modify the clinical course.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Acknowledgments
We thank Dr. Lisa Kratz, Director of Biochemical Genetics of Kennedy Krieger Institute, Baltimore, for her valuable contribution in the diagnostic workup.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Key messages
Unlike other organic acidurias, L2HGA commonly has a chronic progressive clinical course, which often leads to delayed diagnosis Awareness of the characteristic imaging pattern in L2HGA helps in early diagnosis and targeted metabolic and genetic workup Significant proportion of acute encephalopathic presentation and asymmetric imaging findings in our series further expands the clinico-radiological spectrum of L2HGA Good treatment response to riboflavin trial has been described in a proportion of children; early diagnosis thus extends an opportunity to modify the clinical course
REFERENCES
- L-2-hydroxyglutaric aciduria: An inborn error of metabolism? J Inherit Metab Dis. 1980;3:109-12.
- [Google Scholar]
- L-2-hydroxyglutaric acidemia: A novel inherited neurometabolic disease. Ann Neurol. 1992;32:66-71.
- [Google Scholar]
- L-2-hydroxyglutaric aciduria: Pattern of MR imaging abnormalities in 56 patients. Radiology. 2009;251:856-65.
- [Google Scholar]
- An overview of L-2-hydroxyglutarate dehydrogenase gene (L2HGDH) variants: A genotype-phenotype study. Hum Mutat. 2010;31:380-90.
- [Google Scholar]
- L-2-hydroxyglutaric aciduria, a defect of metabolite repair. J Inherit Metab Dis. 2007;30:681-9.
- [Google Scholar]
- L: -2-hydroxyglutaric aciduria, a disorder of metabolite repair. J Inherit Metab Dis. 2009;32:135-42.
- [Google Scholar]
- Plasma lysine concentration and availability of 2-ketoglutarate in liver mitochondria. J Inherit Metab Dis. 2002;25:1-6.
- [Google Scholar]
- A gene encoding a putative FAD-dependent L-2-hydroxyglutarate dehydrogenase is mutated in L-2-hydroxyglutaric aciduria. Proc Natl Acad Sci U S A. 2004;101:16849-54.
- [Google Scholar]
- L-2-hydroxyglutaric aciduria: Identification of a mutant gene C14orf160, localized on chromosome 14q22.1. Hum Mol Genet. 2004;13:2803-11.
- [Google Scholar]
- Progress in understanding 2-hydroxyglutaric acidurias. J Inherit Metab Dis. 2012;35:571-87.
- [Google Scholar]
- Founder effect confirmation of c.241A>G mutation in the L2HGDH gene and characterization of oxidative stress parameters in six Tunisian families with L-2-hydroxyglutaric aciduria. J Hum Genet. 2014;59:216-22.
- [Google Scholar]
- An interesting case of metabolic dystonia: L-2 hydroxyglutaric aciduria. Ann Indian Acad Neurol. 2014;17:97-9.
- [Google Scholar]
- L-2 hydroxyglutaric aciduria presenting with anxiety symptoms. BMJ Case Rep. 2013;2013:bcr2013009512.
- [Google Scholar]
- L-2-hydroxyglutaric aciduria presenting with severe autistic features. Brain Dev. 2008;30:305-7.
- [Google Scholar]
- L-2-hydroxyglutaric aciduria presenting as status epilepticus. Brain Dev. 2001;23:255-7.
- [Google Scholar]
- Hemiconvulsion-hemiplegia-epilepsy syndrome as a presenting feature of L-2-hydroxyglutaric aciduria. J Child Neurol. 2006;21:538-40.
- [Google Scholar]
- L-2-hydroxyglutaric aciduria: Neuropathological correlations and first report of severe neurodegenerative disease and neonatal death. J Inherit Metab Dis. 1996;19:335-43.
- [Google Scholar]
- L-2-hydroxyglutaric aciduria diagnosed in an adult presenting with acute deterioration. J Neurol. 2010;257:146-8.
- [Google Scholar]
- A novel mutation as a cause of L-2-hydroxyglutaric aciduria. J Neurol. 2009;256:672-3.
- [Google Scholar]
- Sudden unexpected death in an infant with L-2-hydroxyglutaric aciduria. Eur J Pediatr. 2009;168:957-62.
- [Google Scholar]
- MRI features in 17 patients with l2 hydroxyglutaric aciduria. Eur J Radiol Open. 2016;3:245-50.
- [Google Scholar]
- Neuroimaging findings of organic acidemias and aminoacidopathies. Radiographics. 2018;38:912-31.
- [Google Scholar]
- L-2 hydroxyglutaric aciduria: Proton magnetic resonance spectroscopy and diffusion magnetic resonance imaging findings. J Comput Assist Tomogr. 2003;27:38-43.
- [Google Scholar]
- Single-voxel MR spectroscopy and diffusion-weighted MRI in two patients with l-2-hydroxyglutaric aciduria. Pediatr Radiol. 2003;33:872-6.
- [Google Scholar]
- L-2-hydroxyglutaric aciduria and brain malignant tumors: A predisposing condition? Neurology. 2004;62:1882-4.
- [Google Scholar]
- Cerebral neoplasms in L-2 hydroxyglutaric aciduria: 3 new cases and meta-analysis of literature data. AJNR Am J Neuroradiol. 2012;33:940-3.
- [Google Scholar]
- Gliomatosis cerebri in L-2-hydroxyglutaric aciduria. Acta Neurol Belg. 2015;115:749-51.
- [Google Scholar]
- Wilms tumor in a child with L-2-hydroxyglutaric aciduria. Pediatr Dev Pathol. 2010;13:408-11.
- [Google Scholar]
- Clinical and pathological study of three Tunisian siblings with L-2-hydroxyglutaric aciduria. Acta Neuropathol. 1994;88:367-70.
- [Google Scholar]
- L-2-hydroxyglutaric aciduria: Clinical, neuroimaging, and neuropathological findings. Arch Neurol. 2005;62:666-70.
- [Google Scholar]
- L-2-hydroxyglutaric aciduria: Clinical heterogeneity versus biochemical homogeneity in a sibship. Neuropediatrics. 1997;28:314-7.
- [Google Scholar]
- Clinical, biochemical and neuroradiological findings in L-2-hydroxyglutaric aciduria. Neurol Sci. 2000;21:103-8.
- [Google Scholar]
- Riboflavin treatment in a case with l-2-hydroxyglutaric aciduria. Eur J Paediatr Neurol. 2009;13:57-60.
- [Google Scholar]
- A successfully treated adult patient with L-2-hydroxyglutaric aciduria. Neurology. 2008;70:1051-2.
- [Google Scholar]




