
Translate this page into:
Chudley–McCullough Syndrome
Address for correspondence: Dr. Meltem Özdemir, Department of Radiology, University of Health Sciences, Dışkapı Training and Research Hospital, Ziraat Mah, Şehit Ömer Halisdemir Cad. No: 20, Altıdağ/Ankara, Turkey. E-mail: meltemkaan99@gmail.com
-
Received: ,
Accepted: ,
This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Chudley-McCullough syndrome (CMS), an autosomal recessive condition first reported by Chudley et al., in 1997, comprises profound sensorineural hearing loss and specific brain abnormalities. The hearing loss may be congenital or early onset. Brain abnormalities are striking, but despite these brain malformations, individuals with CMS do not present significant neurodevelopmental abnormalities. Recently, the cause of CMS has been shown to be the inactivating mutations in G protein signaling modulator 2. We aimed to present a 36-year-old male who has the characteristic clinical and neuroimaging findings of CMS.
Keywords
Cerebellar dysplasia
Chudley-McCullough syndrome
corpus callosum hypogenesis
sensorineural hearing loss


INTRODUCTION
Chudley–McCullough syndrome (CMS) is an autosomal recessively inherited disorder characterized by severe-to-profound sensorineural hearing loss and specific structural brain abnormalities. This syndrome was first recognized in a Canadian–Mennonite consanguineous family in which a brother and sister had hydrocephalus and profound bilateral sensorineural deafness. To date, 21 cases have been reported as having specific features of CMS.[12345678]
Despite striking brain malformations, individuals with CMS do not present significant neurodevelopmental abnormalities, except for hearing loss. The hearing loss may be congenital or early onset.[49] Here, we present a 36-year-old male from a Turkish consanguineous family, with bilateral severe sensorineural hearing loss and characteristic brain malformations.
CASE REPORT
A 36-year-old male with a bilateral sensorineural hearing loss was referred to the Department of Radiology for a brain magnetic resonance imaging (MRI). He is the first of two brothers born to healthy parents of Turkish descent, who are first cousins. The younger brother is healthy. There is no other family history of deafness in the extended family. At the age of 9 months, the hearing impairment was noticed. An audiologic assessment confirmed severe bilateral sensorineural hearing loss, and the loss was corrected with a hearing aid. Hearing loss has remained stable. He had no school problems and graduated from high school. On physical examination, he has no dysmorphic features. He has neither an obvious neurological difficulty nor a cognitive impairment.
A brain MRI study without contrast administration was performed on a 1.5 Tesla scanner (Integra, Philips). Sagittal FLAIR images demonstrated corpus callosum hypogenesis [Figure 1]. The rostrum, genu, and splenic portions of the corpus callosum were absent, and the body part was seen as a short thin line. Lateral ventricles were noted to be slightly small in caliper and slit like in coronal T2-weighted images [Figure 2]. There were bilateral frontal parasagittal polymicrogyria [Figures 1, 3 and 4]. Nodular gray matter heterotopia was present in periventricular regions [Figures 2–4]. In the inferior aspects of both cerebellar hemispheres, there were abnormal foliation and fissuration with loss of the normal architecture, consistent with cerebellar dysplasia [Figure 5]. The findings of temporal computed tomography and contrast-enhanced temporal MRI were both normal. A follow-up brain MRI study has been planned, but not performed yet.
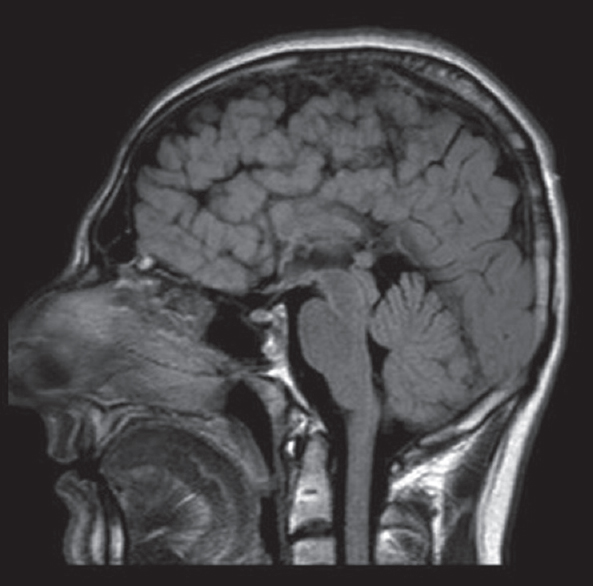
- Sagittal fluid-attenuated inversion recovery image delineates corpus callosum hypogenesis. The rostrum, genu, and splenium are absent while remnant of the body seems as a short thin line. Note polymicrogyria in the frontal lobe.
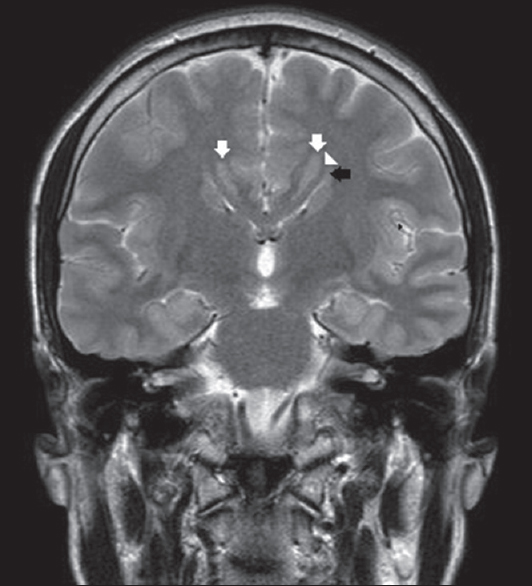
- Corpus callosum is absent in this coronal T2-weighted section through thalami. Lateral ventricles are slit like and small in caliper. They are upturned (black arrow) and there are Probst bundles (arrowhead), secondary to callosal hypogenesis. Note bilateral periventricular nodular gray matter heterotopia (white arrows).
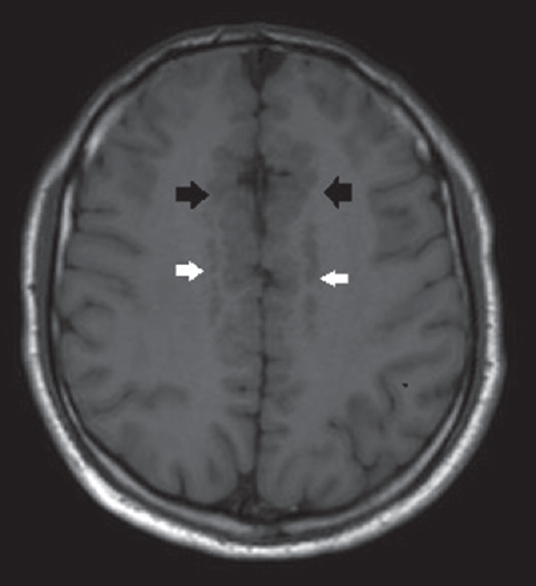
- Axial T1-weighted image shows bifrontal parasagittal polymicrogyria (black arrows) and bilateral periventricular nodular gray matter heterotopia (white arrows).
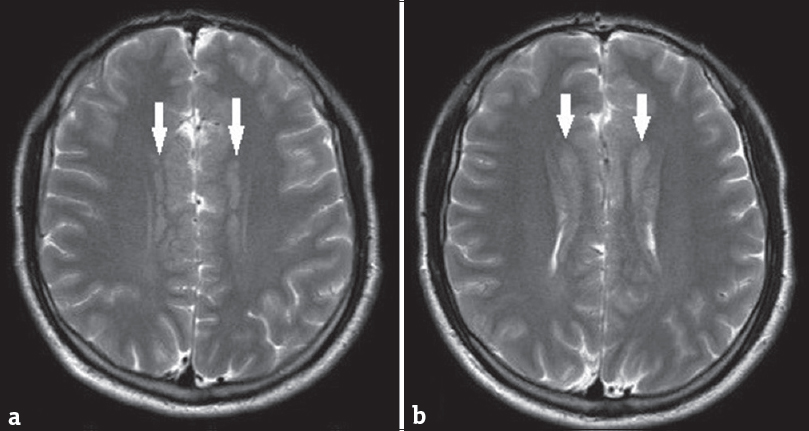
- Subsequent axial T2-weighted images clearly delineate bilateral periventricular gray matter heterotopia (white arrows). Note bifrontal parasagittal polymicrogyria (a and b).
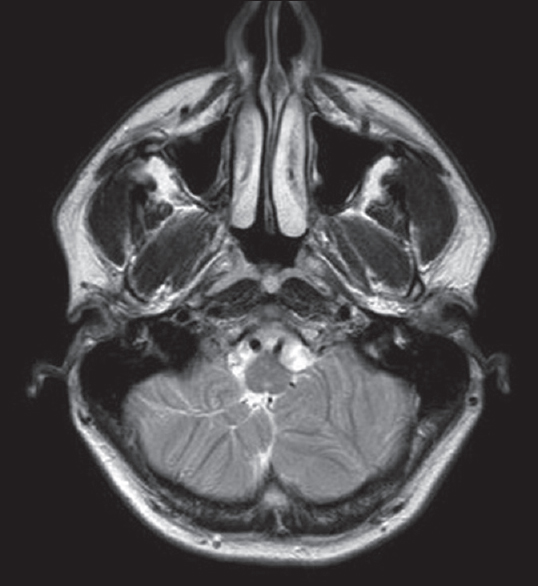
- Axial T2-weighted image demonstrates abnormal cerebellar foliation and fissuration with loss of the normal architecture in the inferior aspect of the cerebellar hemispheres, consistent with cerebellar dysplasia.
Based on the presence of these rather specific neuroimaging findings and the history of bilateral early-onset severe sensorineural hearing loss, the patient was diagnosed as having CMS.
DISCUSSION
After being first described in 1997,[1] several subsequent case reports further delineated the clinical phenotype of CMS. The clinical hallmarks of the syndrome are autosomal recessive inheritance, severe-to-profound sensorineural hearing loss, and partial agenesis of the corpus callosum. Distinctive combination of brain malformation, which is not seen in any other genetic syndromes, includes partial agenesis of the corpus callosum, frontal polymicrogyria, gray matter heterotopia, cerebellar dysplasia, and arachnoid cysts.[9]
In 2012, Doherty et al.[9] identified biallelic mutations in G protein signaling modulator 2 (GPSM2) as the genetic cause of CMS. They investigated individuals with CMS from eight families (5 Mennonite, 1 European-American, 1 Dutch, and 1 Mexican-American family) and identified four deleterious mutations. In this study group, all of the cases had ventriculomegaly and most of them presented mild-to-moderate motor and/or communication delay. Posterior agenesis of the corpus callosum was uniformly present in all cases. Except for the two cases of whom MRI was not available, heterotopia, frontal polymicrogyria, and cerebellar dysplasia were invariable neuroimaging findings in the study group. Nearly all of the cases showed interhemispheric, cerebellopontine, or pineal cysts of variable sizes.
Imaging findings of four additional cases from the two families (1 Palestinian and 1 Turkish family), who had previously been reported as having nonsyndromic hearing loss and truncating mutations in GPSM2,[78] were also evaluated in the study of Doherty et al. All four cases displayed imaging features consistent with CMS. However, in contrast to the other groups, none of them had ventriculomegaly, and corpus callosum abnormalities tended to be less severe. They also differed from the other groups not presenting any neurological deficits. The case we present typically showed the same clinical and neuroimaging features as these four individuals from families of Middle East origin. He had no ventriculomegaly and no neurologic disorder, and he showed the distinctive imaging findings of CMS. Our patient had rejected the genetic counseling, so the genetic nature of the disorder could not be identified.
GPSM2 is necessary for maintenance of cell polarity and spindle orientation, both of which are essential for proper tissue development. The expression of GPSM2 takes place at the apical surfaces of hair and supporting cells of inner air structures and is critical for the early development of hearing.[10] GPSM2 is also required for correct spindle orientation for neurotransmitter localization in mature neurons.[9] It has been clearly shown that inactivating mutations in GPSM2 cause CMS.[6910]
Although they have prominent structural brain abnormalities, patients with CMS do not have significant seizures or cognitive impairment. Therefore, it is recommended for all patients with sensorineural hearing loss, whether or not they have obvious signs of brain dysfunction, to be screened for GPSM2 mutations and undertake a brain imaging. Syndromic forms of corpus callosum abnormalities, including Aicardi, Apert, and Zellweger syndromes, have distinctive clinicopathologic associations which mostly present in neonates with dysmorphic findings, feeding difficulties, and/or seizures. On the other hand, nonsyndromic forms may be asymptomatic and are often associated with highly variable combinations of other central nervous system abnormalities. Therefore, CMS must be kept in mind as a differential in the cases with combined central nervous system abnormalities accompanied by sensorineural hearing loss.[11] GPSM2 sequencing in fetuses and infants with agenesis of corpus callosum and heterotopia is recommended as identification of GPSM2 mutations would greatly alter prognostic counseling and allow for early detection and treatment of hearing loss after birth.[910]
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2018/8/1/45/245525
REFERENCES
- Bilateral sensorineural deafness and hydrocephalus due to foramen of monro obstruction in sibs: A newly described autosomal recessive disorder. Am J Med Genet. 1997;68:350-6.
- [Google Scholar]
- Brothers with Chudley-McCullough syndrome: Sensorineural deafness, agenesis of the corpus callosum, and other structural brain abnormalities. Am J Med Genet A. 2004;124A:74-8.
- [Google Scholar]
- Sensorineural deafness, hydrocephalus and structural brain abnormalities in two sisters: The Chudley-McCullough syndrome. Am J Med Genet A. 2006;140:1183-8.
- [Google Scholar]
- Chudley-McCullough syndrome: Another report and a brief review of the literature. Clin Dysmorphol. 2011;20:107-10.
- [Google Scholar]
- Chudley-McCullough syndrome: Case report and review of the neuroimaging spectrum. Neuropediatrics. 2012;43:44-7.
- [Google Scholar]
- GPSM2 and Chudley-McCullough syndrome: A Dutch founder variant brought to North America. Am J Med Genet A. 2013;161A:973-6.
- [Google Scholar]
- Whole exome sequencing and homozygosity mapping identify mutation in the cell polarity protein GPSM2 as the cause of nonsyndromic hearing loss DFNB82. Am J Hum Genet. 2010;87:90-4.
- [Google Scholar]
- A truncating mutation in GPSM2 is associated with recessive non-syndromic hearing loss. Clin Genet. 2012;81:289-93.
- [Google Scholar]
- GPSM2 mutations cause the brain malformations and hearing loss in Chudley-McCullough syndrome. Am J Hum Genet. 2012;90:1088-93.
- [Google Scholar]
- GPSM2 mutations in Chudley-McCullough syndrome. Am J Med Genet A. 2012;158A:2972-3.
- [Google Scholar]
- Agenesis of the corpus callosum: An MR imaging analysis of associated abnormalities in the fetus. AJNR Am J Neuroradiol. 2009;30:257-63.
- [Google Scholar]




