
Translate this page into:
Bilateral oligodendroglial hamartomas: A rare cause of drug-resistant epilepsy in a pediatric patient

*Corresponding author: Havisha Munjal, Department of Imaging Sciences, University of Rochester Medical Center, Rochester, New York, United States. havisha_munjal@urmc.rochester.edu
-
Received: ,
Accepted: ,
How to cite this article: Munjal H, Mistry DI, Almast J, Ellika S. Bilateral oligodendroglial hamartomas: A rare cause of drug-resistant epilepsy in a pediatric patient. J Clin Imaging Sci 2022;12:24.
Abstract
Intractable or drug-resistant seizures in pediatric patients are often secondary to cortical malformations, hamartomas, or mass lesions. Various subtypes of intracerebral hamartomas, associated with seizure disorders, have been described. In this report, we describe a subtype of intracerebral hamartoma associated with intractable epilepsy in a 10-year-old patient. Initial MR imaging demonstrated a mildly expansile, T2/FLAIR hyperintense, T1 isointense, nonenhancing lesion with blurring of the gray-white junction in the left amygdala. Surgical resection was performed, and pathology confirmed oligodendroglial hamartoma. Patient’s seizures recurred after a two-year interval with imaging demonstrating a similar lesion in the right amygdala which in retrospect was also seen on multiple imaging studies. This case report demonstrates the importance of recognizing oligodendroglial hamartomas as a cause of intractable seizures given the imaging findings, distinguishing it from ganglioglioma, dysembryoplastic neuroepithelial tumor, and oligodendroglioma, and the importance of closely looking/searching for contralateral lesions, which has important therapeutic and prognostic implications.
Keywords
Oligodendroglial hamartoma
Epilepsy
Pediatric

INTRODUCTION
Cerebral hamartomas are lesions composed of disorganized mature neuronal or ganglion cells, glial cells, and blood vessels and are a rare cause of medically intractable focal epilepsy.[1-3]
We describe the MR imaging findings in a subtype of cerebral hamartomas composed of mature oligodendroglial cells, called oligodendroglial hamartoma, in a patient with medically intractable epilepsy. The lesion was initially seen in the left amygdala with discovery of a similar lesion in the contralateral right amygdala after surgical resection on the left. This report highlights the imaging findings of oligodendroglial hamartoma and the need to pay close attention to the contralateral cerebral hemisphere to identify unsuspected lesions on the contralateral side which has important therapeutic and prognostic implications.
CASE REPORT
A 10-year-old, right-handed female patient presented to the Emergency Department with acute-onset complex partial seizures characterized by left eye twitching followed by nausea and emesis. Physical exam was notable for a depressed respiratory rate, disconjugate eye gaze, and a GCS of 7. Electroencephalogram (EEG) demonstrated epileptiform discharges from the left temporal lobe.
The initial CT head was unrevealing. Interictal MR imaging demonstrated an expansile T1 isointense, T2/FLAIR hyperintense, nonenhancing mass lesion with blurring of the gray matter- white matter junction centered in the left amygdala with arterial spin-labeled perfusion imaging demonstrating decreased relative cerebral blood flow/hypoperfusion in the left temporal lobe [Figure 1]. The initial imaging diagnosis was a focal cortical dysplasia with an associated glioneuronal tumor. Clinically, no overgrowth syndromes were suspected. Given the imaging findings and persistent drug-resistant seizures, the patient underwent left temporal craniotomy for resection of the mass lesion. Pathology revealed an oligodendroglial hamartoma [Figure 2].
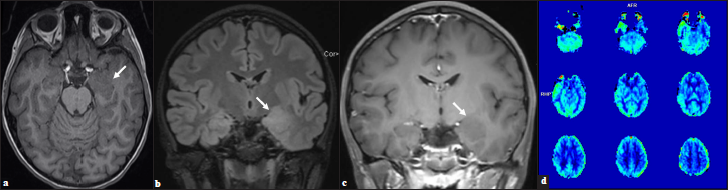
- (a) Ill-defined, expansile, T1 isointense, (b) T2 hyperintense, (c) Nonenhancing lesion within the left amygdala and hippocampus with blurring of the gray-white junction in the adjacent anteromedial temporal lobe (arrows). (d) Perfusion imaging demonstrates relative decrease in cerebral blood flow in the left temporal lobe.
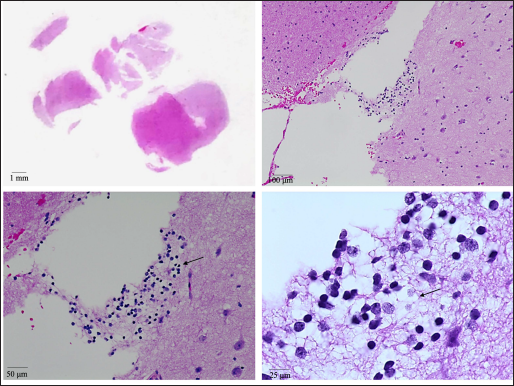
- H&E images (at various magnifications) showing a cluster of oligodendrocytes with round nuclei and cytoplasmic clearings (arrows) in the subcortical white matter. Neurons with larger nuclei (relative to oligodendrocytes) can be seen in the adjacent cortex.
Postoperatively, the patient continued to have intermittent episodes of freezing, increased irritability, and facial twitching without emesis and remained on antiepileptic medications and a benzodiazepine. It was unclear whether these episodes represented an atypical presentation of her seizures. Two years postresection, the patient presented with typical myoclonic seizures associated with confusion, memory loss, and lip-smacking followed by postictal disorientation and emesis. The patient underwent long-term EEG monitoring which revealed seizures localized to the right frontotemporal region with frequent right frontotemporal discharges and additional frequent generalized spike, polyspike discharges suggestive of a possible idiopathic generalized epilepsy trait or an intracortical focus of seizures.
Given these findings, additional imaging was performed. Repeat MR imaging demonstrated a new ill-defined lesion within the right amygdala with similar characteristics as the previously resected left amygdala oligodendroglial hamartoma [Figure 3]. Retrospectively, the right-sided lesion was subtle but noticeable in prior imaging studies including preoperative MR imaging [Figure 4]. After removal of the left-sided oligodendroglial hamartoma, the right-sided lesion became more apparent with imaging characteristics similar to the left-sided oligodendroglial hamartoma, representing a new seizure focus. Given that the lesion was noticeable in multiple prior imaging studies and did not resolve, postictal edema was not considered in the differential. A combination of several anticonvulsants and benzodiazepines is currently being used for successful therapeutic management of seizure activity; however, the recurrence of seizures is hypothesized to be secondary to the right amygdala oligodendroglial hamartoma.
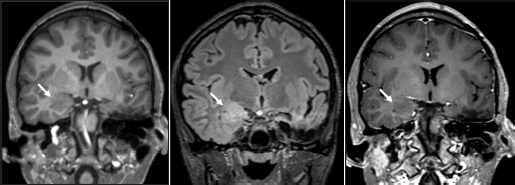
- (a) Status-post left anterior temporal lobectomy and left amygdalohippocampectomy with expected postsurgical changes. New ill-defined T1 isointense, (b) T2 FLAIR hyperintense, (c) and nonenhancing lesion in the right amygdala with blurring of the gray-white junction in the right inferomedial temporal lobe (arrows).
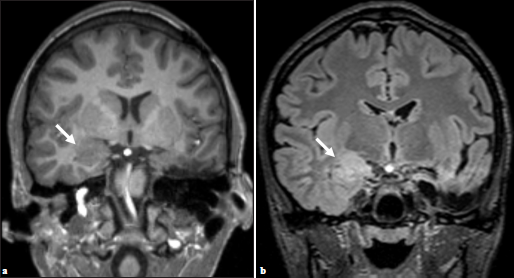
- (a) Preoperative imaging demonstrating subtle, ill-defined T2 hyperintense lesion within the right amygdala, (b) which appears increased in constituency on postsurgical T2 FLAIR (arrows).
DISCUSSION
In this case report, we describe bilateral oligodendroglial hamartomas as the etiology of drug-resistant epilepsy in a pediatric patient. While unilateral cases of oligodendroglial hamartomas have been reported, to our knowledge, no other case(s) of bilateral nonhypothalamic oligodendroglial hamartomas have been described in the literature.
There are multiple etiologies for intractable or drug-resistant seizures in pediatric patients with common structural abnormalities including cortical malformations, tumors, hippocampal sclerosis, and ischemia.[4] Cortical malformations, including focal cortical dysplasia, microgyria, polymicrogyria, and glioneuronal hamartomas, have been identified as a common etiology of intractable seizures in both pediatric and adult populations.[5,6] Approximately 30% of intractable epilepsy is secondary to tumors, and the most commonly encountered tumors are multiple subtypes of low-grade gliomas.[4,7] Of the low-grade gliomas, gangliogliomas are most common with a reported incidence of 37%, followed by dysembryoplastic neuroepithelial tumors with an incidence of 14% and low-grade fibrillary astrocytomas with an incidence of 11%.[4] Other lesions leading to drug-resistant seizures are subtypes of cerebral hamartomas, including glioneuronal hamartomas and oligodendroglial hamartomas.[1,3,8]
Diehl et al. described the imaging characteristics in 14 patients with intracranial hamartomas. The hamartomatous lesions were well-circumscribed, demonstrated a high signal on T2-weighted imaging, iso- or hypointense signal on T1 weighted imaging, and lacked enhancement on postcontrast scans.[1,5] Gray white matter blurring was frequently seen in their cohort of patients with associated cortical dysplasia on pathology.
Pathologically gangliogliomas are composed of two cellular components that are differentiated from glioneuronal hamartomas by increased atypia.[1] Dysembryoplastic neuroepithelial tumors, demonstrate glioneuronal elements with microcystic changes and floating neurons whereas oligodendrogliomas are highly cellular and demonstrate cells that have uniform-appearing nuclei and perinuclear clearing.[1,7] In contrast, oligodendroglial hamartomas are marked by large aggregates of mature, oligodendroglial cells with round central nuclei.[3] Similarly, glioneuronal hamartomas are marked by a bland astrocytic component and a ganglionic component that lack significant atypia.[1,9,10] Additional immunohistochemical labeling of Ki-67 or MIB-1 monoclonal antibodies will be mildly present in low-grade gliomas but are often negative in cerebral hamartomas and other benign lesions.[7,10] These findings are important for the differentiation of hamartomatous lesions from low-grade gliomas and have implications for management.
CONCLUSION
Our case demonstrates the importance of including benign lesions in the differential when presented with a pediatric patient with drug-resistant seizure activity. MR imaging holds a critical role in diagnosis and demonstrates isointense, nonenhancing lesions on T1-weighted images and hyperintense lesions on T2-weighted FLAIR sequences. These imaging findings can mimic malignant neoplasms including low-grade gliomas, and histopathology aids in the diagnosis of such lesions. Interestingly, bilaterality of such benign lesions has not previously been noted and recurrence of benign lesions should be included in the differential when presented with recurrence of symptoms. This case emphasizes the importance of recognizing oligodendroglial hamartomas as a cause of intractable seizures and describes its imaging appearance. Additionally, it is imperative to carefully examine the contralateral side as these lesions can occur bilaterally which can have therapeutic and prognostic implications.
It is important to thoroughly assess the contralateral hemisphere when presented with such lesions initially, as subtle contralateral lesions may be missed and bilaterality can impact treatment options. Surgical resection in such bilateral cases may not result in the resolution of symptoms as demonstrated by this case.
ACKNOWLEDGMENTS
We would like to thank Dr. Rajnish Bharadwaj for his assistance with obtaining the pathology correlation for this case report.
Declaration of patient consent
Patient’s consent not required as patient’s identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Hamartomas and epilepsy: Clinical and imaging characteristics. Seizure. 2003;12:307-11.
- [CrossRef] [PubMed] [Google Scholar]
- Spinal cord hamartoma: Case report. Neurosurgery. 1999;44:1125-7. discussion 1127–8
- [CrossRef] [PubMed] [Google Scholar]
- Oligodendroglial hamartoma of the right temporal lobe: A case report and discussion of possible histogenesis. Clin Neuropathol. 1994;13:204-15.
- [PubMed] [Google Scholar]
- Tumours arising in the setting of paediatric chronic epilepsy. Pathology. 2010;42:426-31.
- [CrossRef] [PubMed] [Google Scholar]
- Imaging and radiological-pathological correlation in histologically proven cases of focal cortical dysplasia and other glial and neuronoglial malformative lesions in adults. Neuroradiology. 2000;42:157-67.
- [CrossRef] [PubMed] [Google Scholar]
- Abnormalities of gyration, heterotopias, tuberous sclerosis, focal cortical dysplasia, microdysgenesis, dysembryoplastic neuroepithelial tumour and dysgenesis of the archicortex in epilepsy. Clinical, EEG and neuroimaging features in 100 adult patients. Brain. 1995;118:629-60.
- [CrossRef] [PubMed] [Google Scholar]
- Low-grade gliomas. Oncologist. 2014;19:403-13.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Oligodendroglial hamartoma: A potential source of misdiagnosis for oligodendroglioma. J Neurooncol. 2011;101:325-8.
- [CrossRef] [PubMed] [Google Scholar]
- Hamartomas in the setting of chronic epilepsy: A clinicopathologic study of 13 cases. Hum Pathol. 1997;28:227-32.
- [CrossRef] [PubMed] [Google Scholar]
- Intraventricular glioneuronal hamartoma: Histopathological correlation with magnetic resonance spectroscopy. J Neurooncol. 2005;74:325-8.
- [CrossRef] [PubMed] [Google Scholar]




