
Translate this page into:
Incidental paraganglioma of sella : A case report and literature review


*Corresponding author: Shota Yoshimura, Department of Neurosurgery, Nagasaki Prefecture Shimabara Hospital, Shimabara, Japan. gm.yy.shota@gmail.com
-
Received: ,
Accepted: ,
How to cite this article: Yoshimura S, Yamaguchi S, Hayashi T, Matsuo T. Incidental paraganglioma of sella: A case report and literature review . J Clin Imaging Sci. 2025;15:16. doi: 10.25259/JCIS_140_2024
Abstract
Most primary paragangliomas of the head and neck occur in the carotid, jugular body, tympanic ventricle, and vagus nerves. Primary sellar paragangliomas are rare, and their long-term outcomes remain unknown. It is also unclear whether they can be classified as asymptomatic incidentalomas in the sellar region. A 75-year-old man who had been followed up for 15 years for an asymptomatic non-functional pituitary adenoma strongly requested surgery and underwent endoscopic transsphenoidal surgery to remove the tumor. Intraoperatively, the tumor was found to be elastic, harder than the pituitary adenoma, fibrous, and not extremely vascularized. The tumor was excised extracapsularly, although residual tumor tissue remained in the medial part of the bilateral cavernous sinuses. A histopathological assessment revealed negative epithelial markers, positive neuroendocrine markers, and partial positivity for S-100, leading to a diagnosis of paraganglioma. Cervicothoracic and abdominal computed tomography, along with spinal magnetic resonance imaging, revealed no apparent neoplastic lesions. The patient experienced no recurrence for 5 years following the resection. The majority of sellar tumors are pituitary adenomas, craniopharyngiomas, Rathke’s cleft cysts, or metastatic brain tumors. Herein, we present a case of an asymptomatic primary sellar paraganglioma that was successfully resected. The case highlights that paraganglioma can be included among incidentalomas in the sellar region. Routine follow-up should generally be recommended for patients with asymptomatic sellar incidentalomas.
Keywords
Paraganglioma
Pituitary incidentaloma
Sellar

INTRODUCTION
Primary paragangliomas of the head and neck often occur in the carotid body (60–70%), jugular body, tympanic cavity, and vagus nerve, and account for only 0.6% of all head-and-neck tumors.[1] The majority of sellar tumors consist of pituitary adenomas, craniopharyngiomas, Rathke cleft cysts, and metastasis.[1] The first case of a suprasellar paraganglioma was reported in 1967.[2] In general, paragangliomas are highly vascular neoplasms, a characteristic commonly observed in those located in the head-and-neck region.[2] Surgical treatment for sellar tumors is typically indicated for patients with symptoms such as headache and visual deficits, leading to a pathological diagnosis. Therefore, it remains unclear whether paragangliomas can be classified as asymptomatic pituitary incidentalomas.
This report presents a rare case of a pituitary incidentaloma due to primary sellar paraganglioma with a good long-term prognosis. We also review the literature on sellar paragangliomas.
CASE REPORT
A 75-year-old male with a history of gastric ulcers, colonic polyps, and cataracts but with no specific family history had been followed up for an asymptomatic, non-functioning pituitary adenoma. This diagnosis followed a brain scan 15 years ago, which identified the pituitary tumor. The patient had no symptoms or tumor growth, but he strongly requested surgery. A tumor resection was performed, and laboratory examinations showed that the pituitary hormone concentrations were within the reference range. The ophthalmological examination findings were normal. Magnetic resonance imaging (MRI) showed a 14 × 19 mm neoplastic lesion extending from the intrasellar to the suprasellar regions without compression of the optic nerve. No invasion of the cavernous sinus or narrowing of the internal carotid artery (Knosp grade 1) was observed. The lesion was isointense on T1-weighted images [Figure 1a], hyperintense on T2-weighted images [Figure 1b], and heterogeneously enhanced on Gd T1-weighted images [Figure 1c]. No flow-void signs were observed on MRI. The preoperative diagnosis was a non-functioning pituitary adenoma, and the tumor was removed through endoscopic transnasal sphenoidal surgery.
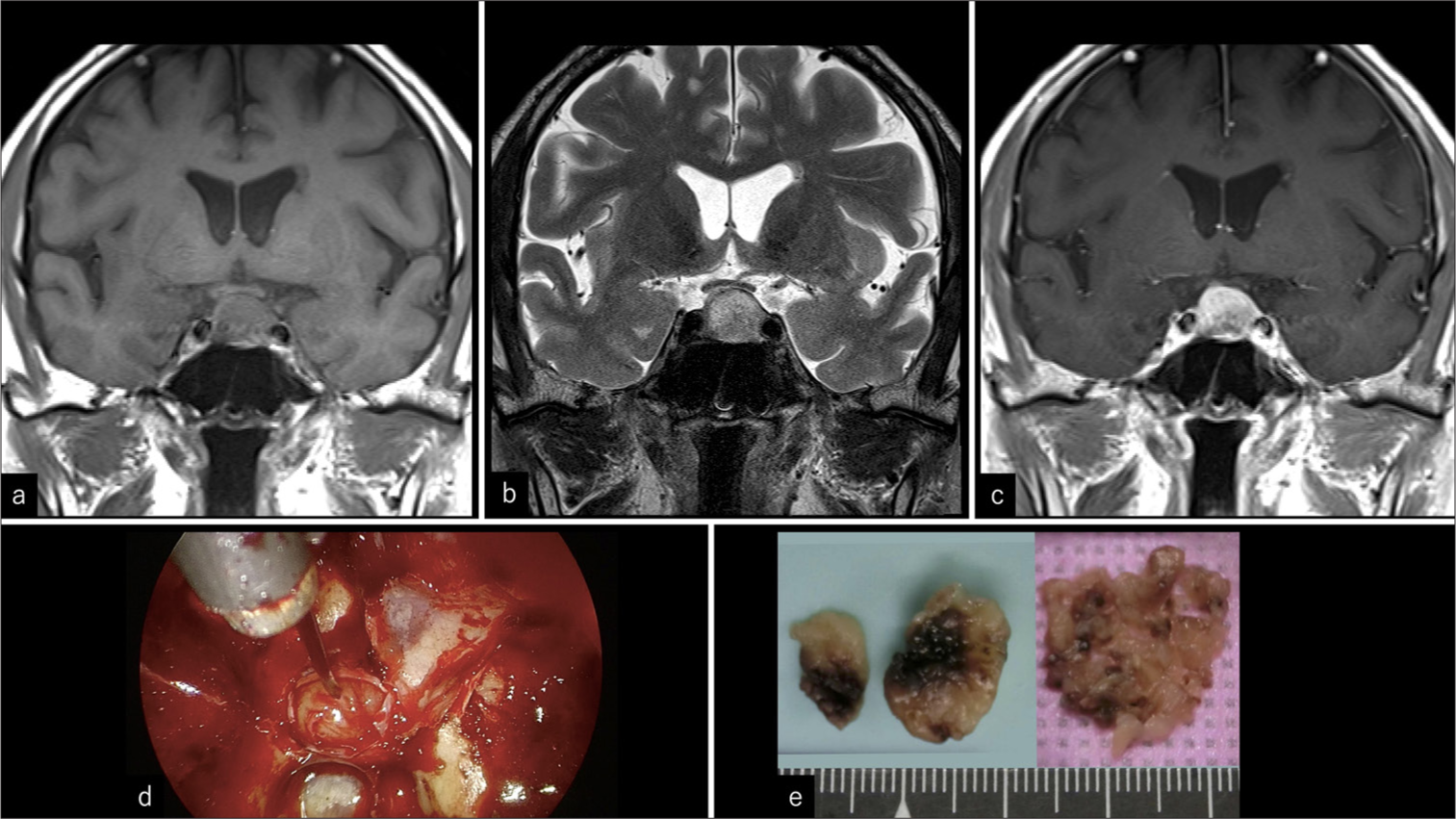
- Magnetic resonance imaging and intraoperative findings. (a) Isointensity on coronal T1-weighted image. (b) Heterogeneously hyperintensity on coronal T2-weighted image. (c) Heterogeneously enhancing lesion on coronal gadolinium-enhanced image (d) Elastic, harder, more fibrous, and less vascular tumor compared to a pituitary adenoma. (e) Hard nodules on the left and small pieces on the right side.
Intraoperative findings revealed that the tumor was elastic, harder than typical pituitary adenomas, fibrous, and not extremely vascularized [Figure 1d]. The tumor was excised extracapsularly, although it remained bilaterally in the medial part of the cavernous sinuses. The mean intraoperative blood volume was 45 mL. The postoperative course was uneventful, and the patient was discharged from our hospital 10 days after the surgery. Macroscopic examination revealed a yellowish-white hard nodule, and small collapsed pieces were removed [Figure 1e]. Histopathological assessment (hematoxylin and eosin staining) revealed that the tumor cells were arranged in a nest-like or cord-like pattern (Zellballen) within a relatively wide fibrous stroma [Figure 2a]. The tumor cells exhibited homogeneous round nuclei and an acidophilic cytoplasm [Figure 2a]. Immunopathological analysis revealed positive neuroendocrine markers chromogranin, synaptophysin, and neural cell adhesion molecule (NCAM). S-100 was partially positive for sustentacular cells at the nest margins [Figure 2b-e]. Epithelial markers (cytokeratin) [Figure 2f] were negative. The analysis of the resected sample was negative for tumor cells or stroma. Glial fibrillary acidic protein (GFAP) was also negative, while the neurofilament was positive. The Ki-67 proliferation index was 0.42%.
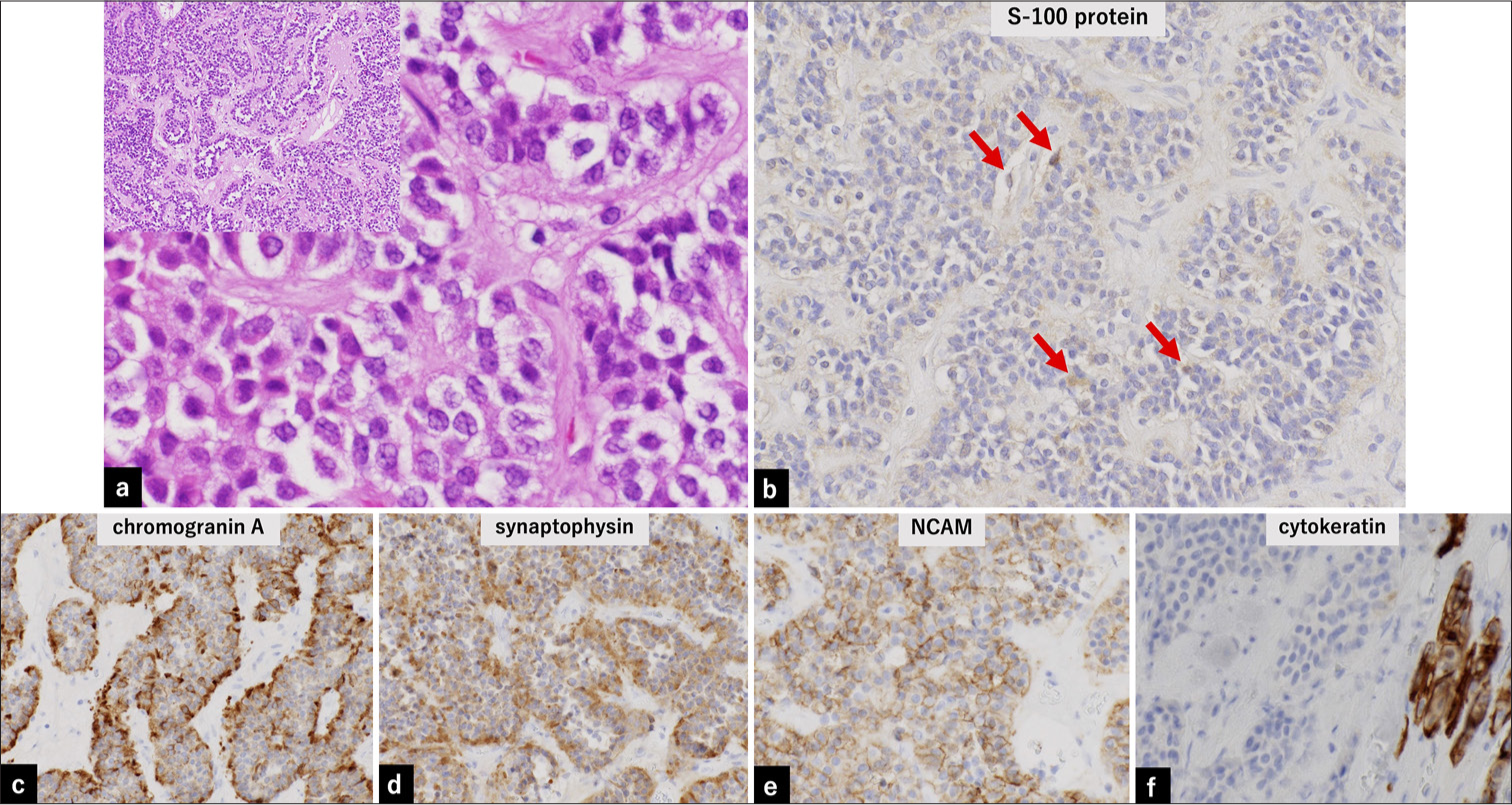
- Pathological findings. (a) Hematoxylin- and eosin-stained histopathology samples. Tumor cells are arranged in a spore-like or cord-like pattern (Zellballen), with a relatively wide fibrous stroma (left upper, ×10). Tumor cells exhibit homogeneous round nuclei and acidophilic cytoplasm (×40 magnification). (b) Immunopathology samples showing partial positivity (red arrow) for S-100 in sustentacular cells at the nest margins and negativity for tumor cells and stroma in the resected sample (×20). (c-e) Positive staining for neuroendocrine markers (chromogranin, synaptophysin, and NCAM) in the resected sample (×20). (f) Negative staining for cytokeratin. The positive area in the lower right represents the normal pituitary gland in the resected sample (×20). NCAM: Neural cell adhesion molecule
These findings led to the diagnosis of paraganglioma. Cervical, thoracic, and abdominal computed tomography and spinal MRI revealed no obvious neoplastic lesions. No recurrence was observed for 5 years after the resection. Informed consent was obtained from the patient for the publication of this manuscript.
DISCUSSION
Paragangliomas are neuroendocrine neoplasms that are composed of paraganglia derived from the neural crest.[3] Paragangliomas predominantly arise in the head-and-neck region, most commonly involving the carotid bodies, the jugular glomus, and vagus nerves, except for the adrenal medulla.[1] Other locations include the larynx, nasopharynx, inferior paraganglionic nodules, nose, orbit, trachea, lungs, heart, and endometrium.[3] Paragangliomas of the head and neck are predominantly asymptomatic, with only 1.2% surgically resected for pathological confirmation of indeterminate masses.[4] The sellar lesions were not included in these cases.[4] Our literature review sought to identify articles on sellar paragangliomas published up to July 1, 2024. We systematically searched the PubMed using the terms “sellar” and “paraganglioma,” and yielded 36 case reports.[1,2,5] Forty reports of sellar, suprasellar, or parasellar paragangliomas indicated an average age of 46.5 ± 21.8 (7–84) years and 60% of the patients were male. The most common symptoms were visual deficits (77.5%) and headaches (62.5%), with no cases reported as asymptomatic. Our case was a pituitary incidentaloma that was followed up for 15 years. In previous reports, the majority of pituitary incidentalomas are classified as pituitary neuroendocrine tumors (PitNETs) and Rathke’s cleft cysts. Pituitary incidentalomas comprise different types of lesions, including neoplastic, cystic, and, non-neoplastic lesions. Sellar paragangliomas were not identified in the autopsy cases.[6] However, the definitions of pituitary tumors or mass lesions differ across reports, with some include only PitNETs among pituitary incidentalomas.[6] The detection frequency of pituitary incidentalomas using imaging is reportedly 5–10%.[6] Pre-operative imaging-based differential diagnosis was challenging and misdiagnosed in 100% of cases.[1,2,5] Two key features have been identified to differentiate sellar paragangliomas from non-functional pituitary adenoma. First, endocrine function is typically normal despite the large tumor size. Second, high vascularization in sellar paragangliomas leads to heterogeneous intensity on T2-weighted images. This is similar to the “salt and pepper” appearance frequently observed in peripheral paragangliomas.[1] The incidentalomas cases followed up may have included those with imaging features of paragangliomas. Furthermore, paragangliomas are encapsulated tumors with rich vascularization, which is reflected by the flow-void sign on MRI.[2] Flow voids are observed in only 20% of the cases, including the no-information cases. Noble et al. reported that this findings were likely due to their predominance in the small dorsal clival meningeal branches of the meningohypophyseal trunk, rather than in the larger vessels such as the ascending pharyngeal artery, typically seen in jugular paragangliomas.[7] Flow voids were not mentioned in most cases, and further exploration of their relationship with vascularity is required. Intraoperatively, the characteristics of the tumor are different from those of a pituitary adenoma, suggesting the presence of an unusual tumor in the sellar region.
The management of pituitary incidentalomas has been established based on the natural history of PitNETs. However, the duration of follow-up has not been established. The decision to perform surgery is often based on individual patient factors such as symptoms, clinically significant growth, loss of endocrinological function, proximity to the optic chiasm, or persistent headaches.[6] The surgery rate for asymptomatic incidentaloma has not been established. The asymptomatic group that underwent surgery for pituitary incidentaloma showed better results in terms of postoperative residual tumors and 5-year recurrence-free rates.[6] As general practice, follow-up is recommended for asymptomatic patients with sellar incidentalomas. Most surgical procedures for symptomatic cases have been performed using transcranial (55%) or transsphenoidal (50%) approaches.[1,2,5] Postoperative radiotherapy is controversial because the natural history of paragangliomas remains unknown. However, postoperative radiotherapy has been used for cases where the tumor has not been completely removed. Adjuvant therapy included radiotherapy and hormone therapy in 52.5% and 5% of the patients, respectively.[1,2,5]
Paragangliomas have also been reported in organs where paraganglial cells are generally absent, such as the sellar region. Two theories have been proposed for the development of paraganglioma. The first suggests that these tumors in the head and neck originate from remnants of neural crest tissue, while the second theory proposes that the paraganglion cells may migrate into the cavernous sinus through the tympanic or ciliary branches of the glossopharyngeal nerve in large tumors extending into the cavernous sinus.[1] Bilbao et al. postulated that the neural crest may be involved in normal pituitary gland development, and a nest of paraganglionic tissue may have been included in the developing adenohypophysis.[8] This is similar to the present case.
The outcomes of sellar paragangliomas vary significantly. Some cases of paragangliomas are characterized by rapid tumor growth, multiple anatomic region invasion, and distant metastasis development. However, the criteria for diagnosing malignancies are controversial. Histopathological features such as central necrosis, hypervascularity, or increased mitotic activity are insufficient to reliably distinguish benign and malignant tumors.[9] The most widely accepted criterion is the presence of local or distant metastases to non-endocrine tissues.[3] Based on this definition, 10% (4 of 40 cases) of all reported cases of sellar paragangliomas have been classified as malignant.[1,2,5] Malignant paragangliomas of the head and neck have been reported for 6% of carotid body paragangliomas, 5% of jugulotympanic paragangliomas, 10–19% of vagal paragangliomas, 3% of laryngeal paragangliomas, and 17% of sinonasal paragangliomas.[10] Thus, the percentage of malignant sellar paragangliomas was comparable to those at other sites. The case discussed in the present study was a primary sellar paraganglioma that had been followed up for 15 years and showed no recurrence for 5 years after resection. The prognosis of our patient appeared favorable, but it is crucial to analyze additional cases to determine the prognostic factors.
CONCLUSION
We discussed a rare case of an asymptomatic primary sellar paraganglioma with a review of the relevant literature. The fact that paragangliomas is included in incidentaloma was clarified. Follow-up is generally recommended for sellar incidentalomas in asymptomatic patients.
Ethical approval
The Institutional Review Board approval is not required.
Declaration of patient consent
The authors certify that they have obtained all appropriate patient consent.
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
Financial support and sponsorship: Nil.
References
- Case report: Paraganglioma in the sellar region: Longitudinal observation and surgical outcome. Front Oncol. 2023;13:1090615.
- [CrossRef] [PubMed] [Google Scholar]
- Sellar-suprasellar paraganglioma: Report of 2 cases and review of literature. World Neurosurg. 2020;140:293-300.
- [CrossRef] [PubMed] [Google Scholar]
- Head and neck paragangliomas: An update on the molecular classification, state-of-the-art imaging, and management recommendations. Radiol Imaging Cancer. 2022;4:e210088.
- [CrossRef] [PubMed] [Google Scholar]
- Benign paragangliomas: Clinical presentation and treatment outcomes in 236 patients. J Clin Endocrinol Metab. 2001;86:5210-6.
- [CrossRef] [PubMed] [Google Scholar]
- Primary sellar paraganglioma: Case report with literature review and immunohistochemistry resource. World Neurosurg. 2019;125:32-6.
- [CrossRef] [PubMed] [Google Scholar]
- An overview of pituitary incidentalomas: Diagnosis, clinical features, and management. Cancers (Basel). 2022;14:4324.
- [CrossRef] [PubMed] [Google Scholar]
- Intrasellar paraganglioma associated with hvpopituitarism. Arch Pathol Lab Med. 1978;102:95-8.
- [Google Scholar]
- Recurrent primary intrasellar paraganglioma. Case Rep Otolaryngol. 2020;2020:2580160.
- [CrossRef] [PubMed] [Google Scholar]
- Head and neck paragangliomas: An overview. Otolaryngol Clin North Am. 2001;34:829-36, v.
- [CrossRef] [PubMed] [Google Scholar]




