
Translate this page into:
Role of Imaging and Cytogenetics in Evaluation of DiGeorge Syndrome - A Rare Entity in Clinical Practice
Address for correspondence: Dr. Rajoo Ramachandran, Department of Radiology, Sri Ramachandra University, Porur, Chennai - 600 116, Tamil Nadu, India. E-mail: drrajoor@gmail.com
-
Received: ,
Accepted: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
DiGeorge syndrome is a congenital genetic disorder that affects the endocrine system, mainly the thymus and parathyroid glands. The syndrome produces different symptoms, which vary in severity and character between patients. It manifests with craniofacial dysmorphism and defects in the heart, parathyroid, and thymus. Patients can present with a palatal deformity and nasal speech. This rare entity is caused mainly due to deletion of chromosome 22q11.2. Radiographic evaluation of DiGeorge syndrome is necessary to define aberrant anatomy, evaluate central nervous system, craniofacial abnormalities, musculoskeletal system, and cardiothoracic contents. It also helps in planning surgical procedures and surgical reconstructions. We report a case of DiGeorge syndrome in a 4-month-old neonate and discuss the clinical, imaging, and cytogenetic findings that helped in the diagnosis of this rare entity.
Keywords
Endocrine
hypocalcemia
thymus

INTRODUCTION

DiGeorge syndrome (DGS) is a disease of the immune system. A patient with this condition is prone to develop infections due to poor T-cell formation and function. DGS is a lifelong condition whose symptoms can be seen in infants and children. This rare syndrome results in hypocalcemia arising from parathyroid hypoplasia, thymic hypoplasia, and outflow tract defects of the heart. DGS mainly results from deletion of chromosome 22q11.2. In this case report, we discuss the radiological features, laboratory findings, and the cytogenetic results that aided in the diagnosis of this rare syndrome.
CASE REPORT
A 4-month-old infant presented with complaints of cough, cold, and breathlessness that had persisted for 2 days. The baby's oral intake had been very poor the earlier day. Initial clinical examination revealed bilateral wheeze on auscultation and subtle dysmorphic facial appearance which included small chin, small mouth, and coarse facial features. The mother gave history of uneventful postnatal period. The baby had received all the necessary vaccines required for her age. The mother also gave history of abortion of her previous pregnancy during the 2nd trimester because of cardiac anomaly identified on fetal ultrasound (USG) scan. In view of its difficulty in breathing, the baby was admitted to the intensive care unit (ICU) and was started on oxygen and nebulization. Nasopharyngeal aspirate showed positivity for respiratory syncytial virus (RSV) bronchiolitis. Blood gas analysis showed respiratory acidosis. In view of respiratory distress and acidosis, the baby was intubated and started on synchronized intermittent mandatory ventilation (SIMV) mode. Blood cultures were negative. Echocardiogram (ECHO) was done and found to be normal. Chest radiograph frontal projection showed bilateral air space opacities with right upper lobe consolidation suggestive of pneumonia [Figure 1]. The baby was started on antibiotics and oral Ribavirin. During the next day in ICU, the child had seizures. Biochemistry analysis showed reduced calcium level of 5.7 mg/dl (normal value: 8.5–10.1 mg/dl). 25-Hydroxy-vitamin D level was low and measured 13.4 ng/ml (normal value: <20 ng/ml). The parathyroid hormone (PTH) level was normal in spite of the low calcium. The baby was started on 1,25-dihydroxycholecalciferol and calcium. Blood investigations showed low hemoglobin measuring 7.5 g/dl (normal value: 11–14 g/dl) and a total WBC count of 10,700 cells/ml. Polymerase chain reaction (PCR) blood analysis showed positivity for cytomegalovirus (CMV). Flow cytometery of lymphocytes showed combined T-cell and B-cell immunodeficiency. In spite of good antibiotic cover and physiotherapy, the right upper lobe consolidation was persistent. Intravenous immunoglobulin and steroids were started to treat persistent infection, following the hematologist's advice. Computed tomography (CT) of the chest with intravenous contrast showed hypoplastic thymus [Figure 2a] and right upper lobe consolidation [Figure 2b]. CT chest with maximum intensity projection (MIP) and volume-rendered (VR) images showed aberrant right subclavian artery [Figure 3a and b]. In view of dysmorphic facial features, chest infection, hypoplastic thymus, immunodeficiency, hypocalcemia, and decreased vitamin D, with a previous history of abortion of the fetus because of cardiac anomaly, a strong possibility of DGS was suspected. Fluorescent in situ hybridization (FISH) study was done to look for chromosomal defects. The results showed deletion of chromosome 22q11.2 [Figure 4], which confirmed the diagnosis of this rare entity. The baby's respiratory distress gradually settled and the infant was discharged on the parent's request.
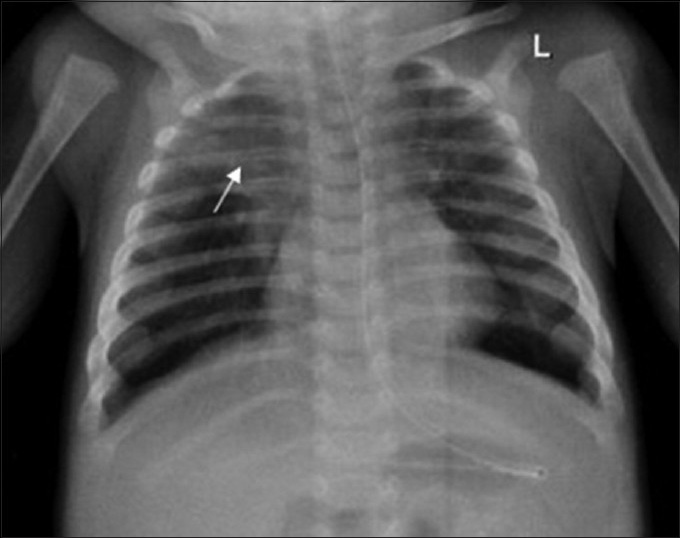
- 4-month-old infant with cough and expectoration, diagnosed with DiGeorge syndrome. Frontal radiograph shows consolidation involving the right upper zone (arrow).
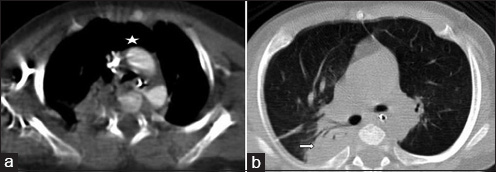
- 4-month-old infant with cough and expectoration, diagnosed with DiGeorge syndrome. (a) Contrast-enhanced axial image of CT chest in mediastinal window shows absent thymus (asterisk). (b) Non-contrast axial image of CT chest in lung window shows consolidation involving the right upper lobe (right arrow).
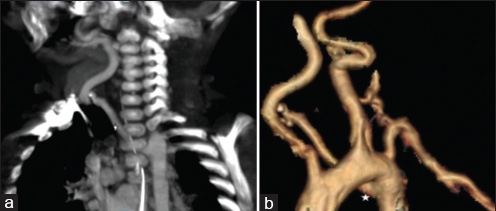
- 4-month-old infant with cough and expectoration, diagnosed with DiGeorge syndrome. (a) CT scan-coronal maximum intensity projection (MIP) in arterial phase of chest shows aberrant right subclavian artery (asterisk). (b) CT scan volume-rendered image shows aberrant right subclavian artery (asterisk).
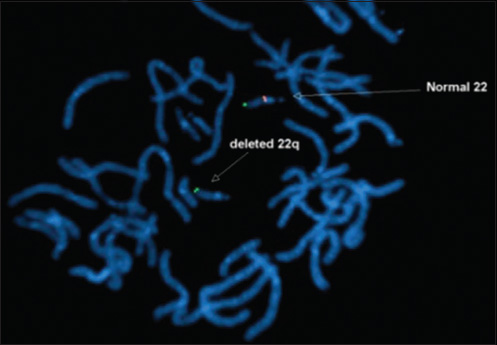
- 4-month-old infant with cough and expectoration, diagnosed with DiGeorge syndrome. Fluorescent in situ hybridization (FISH) study shows deletion of chromosome 22q11.2 (arrow)
DISCUSSION
DGS is a primary immunodeficiency disease caused by deletion of genetic material from chromosome 22q11.2.[1] As part of this genetic defect, the thymus gland may be affected and this impairs the production of T-lymphocytes, which causes recurrent infections. The syndrome was first described by Dr. Angelo DiGeorge in 1965.[2] The incidence of this rare syndrome was around 1 in 5000 births in the general population in the UK, according to the report of Alison et al.[3] More than 180 anomalies have been reported, so far; the expression of DGS varies from person to person and a single affected individual will not have all the anomalies. Other names of DGS include velocardiofacial syndrome and conotruncal anomaly face syndrome.
The pathogenesis behind this rare entity is deletion of chromosome 22q11.2, which causes third and fourth branchial pouch defects and may lead to hypoplasia or absence of thymus and/or parathyroid gland.[4] DGS, in most instances, occurs sporadically and around 10% of the cases are inherited from one of the family members. The defective gene is autosomal dominant and there is 50% risk of transmitting the deletion to the child.[5] However, in our case, the cause is unknown.
The clinical features requiring the clinician to perform 22q11.2 deletion studies may vary from person to person depending on the age of the patient. The common classic findings include conotruncal cardiac anomalies (i.e., ventricular septal defect, tetralogy of Fallot, vascular rings, and interrupted aortic arch), palatal abnormalities (cleft palate and cleft lip), nasal regurgitation and/or hyper nasal speech, behavioral problems, psychiatric illness (bipolar disorder or schizophrenia),[6] immunodeficiency (reduced T lymphocytes leading to recurrent infections),[7] hypocalcemia, and characteristic facial features.[89]
Radiological evaluation of a patient with suspected DGS includes evaluation of central nervous system, craniofacial abnormalities, musculoskeletal system, and cardiothoracic contents. A detailed evaluation of craniofacial system has to be done to check for external auditory canal (EAC) stenosis, prominent nose, thin upper lip, and asymmetric face. CT with contrast of the head and neck is required to identify the presence of aberrant course of internal carotid arteries, hypoplastic adenoids, and middle ear anomalies.[10] Contrast-enhanced CT (CECT) of the chest should be done to check for ventricular septal defect, tetralogy of Fallot, aortic arch anomalies, vascular ring, absent thymus, and to identify lung infections.
It is estimated that approximately 90% of patients with the clinical diagnosis of DGS have a deletion of a specific portion of chromosome number 22 at position 22q11.2, called a microdeletion. This microdeletion is identified by a blood test called FISH analysis. The FISH test has made the diagnosis of DGS more precise and accurate. Approximately 90% of 22q11.2 deletions occur spontaneously and have not been passed on from the mother or father of the child. But once the diagnosis has been made, genetic counseling is critical and testing should be offered to parents and other family members.
Managing DGS needs a multidisciplinary approach and the cause for each symptom should be identified and treated accordingly. In our case, there was thymic hypoplasia (as evident in CT imaging) with decreased T lymphocyte production which, in turn, caused the B lymphocytes to produce insufficient antibodies; hence, immunoglobulin therapy and antibiotics were administered. Blood investigations revealed low calcium and vitamin D levels; appropriate calcium and vitamin D supplements were administered.
CONCLUSION
DiGeorge Syndrome (DGS) is a rare genetic disorder which can cause a wide array of symptoms. The diagnosis is often missed or delayed because pregnant women are not routinely screened for this rare entity. Hence, in any neonate with congenital hypocalcemia, recurrent infection, and seizure with absent parathyroid and thymus gland evident on imaging, a suspicion of DGS should arise and a detailed radiological, clinical, and cytogenetic examination should be carried out to avoid unnecessary complications.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2015/5/1/4/150445
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- Velo Cardio Facial Syndrome or 22q11.2 Deletion Syndrome. An Introduction for Teachers and Carers. VCFS 22q11 Foundation. Available from: http://www.vcfsfa.org.au
- [Google Scholar]
- Discussions on a new concept of the cellular basis of immunology. J Paediatr. 1965;67:907.
- [Google Scholar]
- Seizure and VSD in 2 Month Old Infant. Radiology Cases in Pediatric Emergency Medicine. Vol. 2, Case 2. In: John A, ed. Burns School of Medicine. Vol 2. Honolulu, Hawaii, USA: University of Hawaii; Case 2
- [Google Scholar]
- DiGeorge Syndrome. Children Hospital of Wisconsin. Available from: http://www.chw.org
- [Google Scholar]
- Psychiatric disorders and intellectual functioning throughout development in velocardiofacial (22q11.2 deletion) syndrome. J Am Acad Child Adolesc Psychiatry. 2009;48:1060-8.
- [Google Scholar]
- Chromosome 22q11.2 deletion syndrome: DiGeorge syndrome/velocardiofacial syndrome. Immunol Allergy Clin North Am. 2008;28:353-66.
- [Google Scholar]
- Presenting phenotype in 100 children with the 22q11 deletion syndrome. Eur J Pediatr. 2005;164:146-53.
- [Google Scholar]
- Recognizing a common genetic syndrome: 22q11.2 deletion syndrome. CMAJ. 2008;178:391-3.
- [Google Scholar]
- Syndromes of the first and second branchial arches, part 2: Syndromes. AJNR Am J Neuroradiol. 2011;32:230-7.
- [Google Scholar]




