
Translate this page into:
Localized Cystic Disease of the Kidney: A Rare Cause of Hypertension in a Young Adult
Address for correspondence: Dr. Aynur Solak, Sifa University Hospital, Department of Radiology, Fevzipasa Boulvard, 172/2, 35240, Basmane, Izmir, Turkey. E-mail: aynursolak@yahoo.com
-
Received: ,
Accepted: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Localized cystic disease of kidney (LCDK) is a rare, non-familial, non-progressive renal disorder that is not associated with cysts or disorders in other organs. Only a few cases have been reported in the literature. While this condition is morphologically identical to the autosomal dominant form of polycystic kidney disease, it is not inherited and is not associated with significant deterioration of renal function. We present a case of a 16-year-old male patient who suffered from hypertension for over two years. On imaging we found several, variable-sized cysts in the upper half of the right kidney. The left kidney and lower segment of the right kidney were normal. Selective renal vein catheterization and sampling showed markedly elevated renin level in the right upper segmental vein (92 pg/ml, normal value: 11-33 pg/ml). The patient underwent a right upper heminephrectomy and histopathology was suggestive of LCDK. After surgery, the patient's blood pressure returned to normal levels without any need of antihypertensive medication and he is under follow-up on outpatient basis for the past two years.
Keywords
Computed tomography
cysts
kidney
localized cystic disease of kidney
magnetic resonance imaging
ultrasound


INTRODUCTION
Localized cystic disease of the kidney (LCDK) is rare, benign condition and first described in 1964 as unilateral polycystic renal disease.[1] It is a non-familial, non-progressive renal disorder that is not associated with cysts or disorders in other organs, and it is not related to other genetic cystic diseases. The disease presents as a segmental cystic abnormality in one kidney and is morphologically identical to autosomal dominant polycystic kidney disease (ADPKD).[123] We present a case of LCDK of the right kidney associated with hypertension and ipsilateral flank pain. The clinical, ultrasonographic (US), laboratory (including renin levels of the right and left renal veins), computed tomography and magnetic resonance imaging (MRI) findings of the case have been described in this report. The differential diagnosis and possible pathogenic mechanism are discussed.
CASE REPORT
A 16-year-old male presented with a right abdominal and groin pain. There was no gross hematuria, fever, or lower urinary tract symptoms. Palpation of his right flank area was normal. He had hypertension for 2 years and his blood pressure (BP) measured 190/100 mm Hg at presentation. He had been taking antihypertensive drugs including valsartan hydrochlorothiazide (160/12.5 mg capsule once daily). No other abnormalities were found in the patient's history and on physical examination. Family screening was performed and no abnormalities were found. There was no family history of renal failure or cerebrovascular accident. His chest radiography and electrocardiography findings, urinanalysis, blood urea nitrogen, and serum creatinine levels were within normal limits. Other laboratory tests including lipid profile and liver functions were with in the normal range. His body mass index (BMI) was also in normal range (BMI: 22.1).
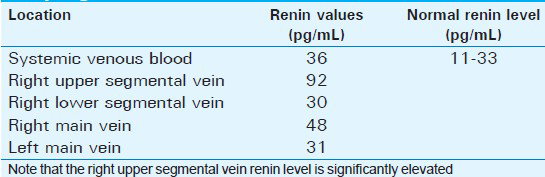
An abdominal ultrasound showed multiple variable-sized cysts in the upper pole of the right kidney with normal left kidney and lower pole of the right kidney [Figure 1]. Renal arterial color Doppler ultrasonography was normal. Abdominal computed tomography confirmed the ultrasound findings that the upper pole of the right kidney was filled with multiple cysts of different sizes with enhancing normal renal tissue between the cysts. There were no cysts in the other intra-abdominal organs. A renin blood test showed that the plasma renin level was moderately elevated (46 pg/ml, normal limits: 11-33 pg/ml). Selective intrarenal catheterization was performed to obtain venous blood samples. His drug was withdrawn four days before the selective renal vein studies and he was given 10 mg furasemide orally in the afternoon prior to the study. The procedure was started after placing the patient in a supine position for at least two hours. A catheter was introduced through the femoral vein and it was advanced using fluoroscopic guidance. Blood samples for plasma renin level were drawn from both main renal veins and from the upper and lower segmental draining veins of each kidney. The highest renin level was measured in the right upper segmental vein (92 pg/ml). Renin levels of the right lower segmental vein and the left renal veins are shown on Table 1.
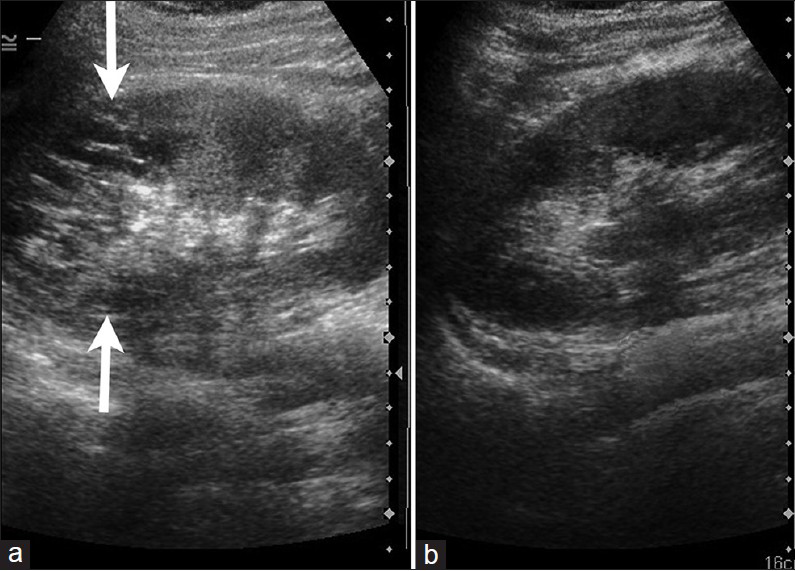
- 16-year-old male with high blood pressure for over 2 years diagnosed with localized cystic disease of the kidney. Ultrasonography of the upper abdomen demonstrates multiple variable-sized cysts in the upper pole (arrows) of the right kidney (a) while the left kidney appears normal (b).
In order to further characterize the lesions, MRI was performed. A gadolinium-enhanced MRI revealed that the upper pole of the right kidney was completely filled with multiple round, well-marginated cysts of varying size without capsule formation [Figure 2]. There were no solid areas within the cysts and renal parenchyma between the cysts was normally enhancing. Technetium-99m-dimercaptosuccinic acid (Tc-99m DMSA) renal scintigraphy showed decreased renal uptake at the upper half of the right kidney. After discussion with the physicians and the parents, a decision was made to perform partial nephrectomy to eliminate the source of the apparent renin-dependant hpertension. The patient underwent right upper pole heminephrectomy with a histopathological result confirming LCDK. Nephrectomy specimen on macroscopic examination showed conglomerate cysts of various sizes in the kidney [Figure 3a]. Microscopy examination showed cysts containing dark brown or clear serous fluid and surrounded by mono-layered flat-cuboidal epithelium without evidence of tumor or papillary formation [Figure 3b].
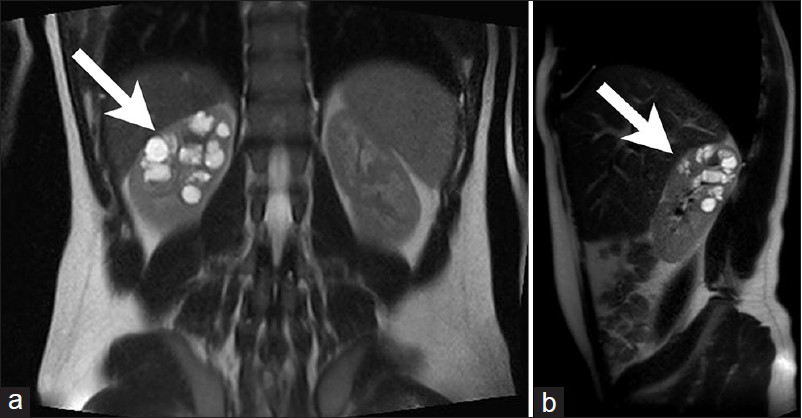
- 16-year-old male with high blood pressure for over 2 years diagnosed with localized cystic disease of the kidney. T2 weighted (a) coronal and (b) sagittal MR images show the upper pole of the right kidney is filled with multiple, round, well marginated cysts of various sizes without capsule formation (arrow).
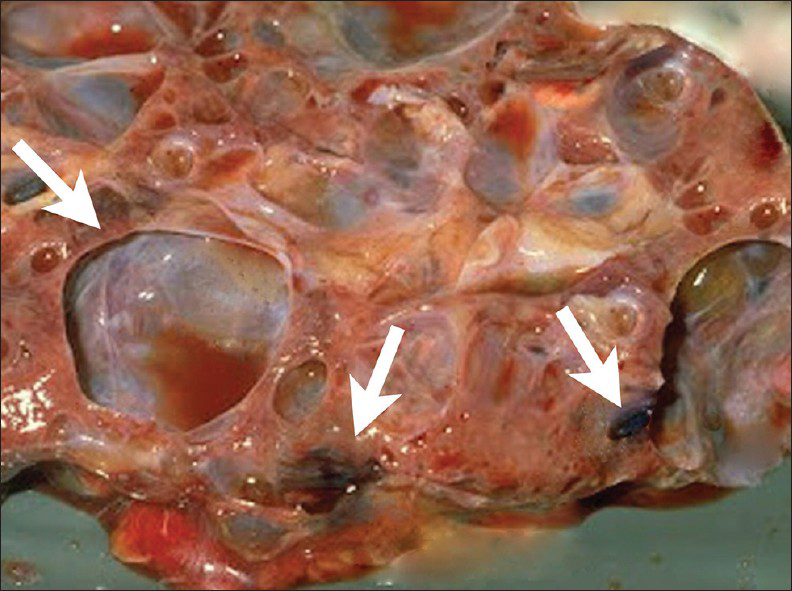
- 16 year old male with high blood pressure for over 2 years diagnosed with localized cystic disease of the kidney. Gross image of the partial nephrectomy specimen shows multiple well demarcated cysts of various sizes (arrows). They contain dark brown or clear serous fluid.
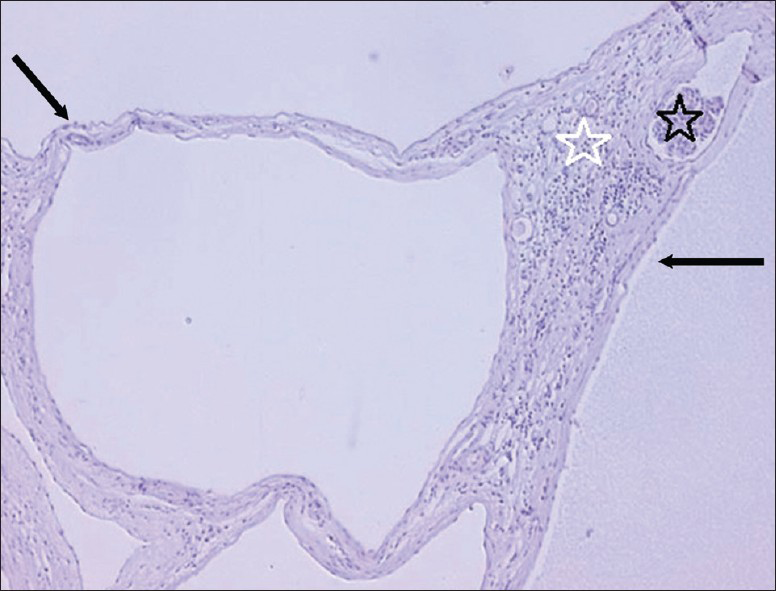
- 16 year old male with high blood pressure for over 2 years diagnosed with localized cystic disease of the kidney. Periodic acid schiff stained (×10) nephrectomy specimen reveals cysts lined largely by a simple low is cuboidal epithelium (black arrows). There are no papillary areas. Hemorrhage noted in the cyst (black star) and inflammatory reaction in which macrophages are prominent is also noted, the intervening tissue between the cysts (white star) contains compressed glomeruli and tubules. The tubules are mature and no blastema or immature fetal elements are identified.
After surgery the patient's blood pressure returned to normal levels without any need for antihypertensive medication. The patient is under follow-up on outpatient basis and has maintained good renal function during the last two years. Follow-up ultrasound examinations showed normally appearing residual right kidney and left kidney.
DISCUSSION
LCDK, first described in 1964 as ‘unilateral polycystic renal disease’, is a rare and poorly understood condition.[1] LCDK is also sometimes called multiple unilateral renal cysts, segmental polycystic kidney disease, and unilateral segmental polycystic renal disease.[123] Kohno and Yunoki reported two patterns of unilateral cystic renal disease involvement as noted on imaging studies. In the diffuse form, multiple cysts are scattered throughout the affected kidney, and in the segmental form, as in our patient, the cysts predominate in one region of the involved kidney.[4] However, in either form, no distinct encapsulated renal mass is formed in contrast to multilocular cystic nephroma.[56] The case series of Slywotzky and Bosniak consisting of 18 cases of localized cystic renal disease, 15 (83%) were males and three were women; the median age at diagnosis was 50 years (range, 24-83 years).[7]
Multiple simple renal cysts and unilateral multicystic dysplastic kidney disease must be considered in the differential diagnosis of the patients with LCDK.[8] In unilateral multicystic dysplastic kidney disease, the kidney is usually non-functioning, as the collecting system is usually atretic or obstructed. While in LCDK, contrast excretion is seen in the collecting system which may be appearing normal or show displacement. Sometimes, multiple simple cysts may be difficult to distinguish from LCDK when the cysts are confined to one segment of the kidney, but they are less numerous than in LCDK. Furthermore, unlike LCDK where the cysts involve both the cortex and the medulla, they are predominantly located in the renal cortex.[1278] In our patient, we observed a normally enhancing renal parenchyma between the cysts in the affected area of the right kidney. The evenly distrubition of cysts in both the renal cortex and medulla and the presence of normally enhancing intervening parenchyma led to the diagnosis of LCDK.
The pathogenesis of LCDK is unclear, although it has been hypothesized that it is due to a developmental error. Gouldesbrough et al., reported that the most important etiological factor is a somatic mutation in the autosomal dominant polycystic kidney gene in the affected segment of the kidney.[9] Sometimes simple cysts can appear in the contralateral kidney simultaneously or several years after the initial diagnosis. Then it becomes indistinguishable from the autosomal dominant polycystic kidney disease both radiologically and pathologically. However, LCDK has five features that are different from ADPKD: 1. Unilateral-partial localization, 2. Negative family history, 3. Absence of progression to chronic renal failure, 4. Absence of cysts in other intra-abdominal organs such as liver, or pancreas, and 5. Absence of disorders affecting other organ systems (colonic diverticuli, cerebral Berry aneurysms, etc.).[1345] Most of these patients present with one or a combination of the following: hypertension, flank pain, hematuria or a flank mass. In our case, family screening was negative for a renal disease and hypertension. But our patient had hypertension and right flank pain.
The mechanism of hypertension in LCDK is similar to that of ADPKD. Renin-containing cells are present in the walls of the cysts and in cells of the connective tissue surrounding the cysts. Renin can also be produced by the epithelial cells lining the cysts and active renin is often present within the cyst fluid.[810] Blood obtained from the main renal vein and segmental renal vein in this study contributed useful information and permitted localization of sources of renin production. Based on the identification of the source of renin production, we recommended right heminephrectomy. Indeed, removal of the upper lobe including multiple various sized cysts led to normalization of the patient's blood pressure.
CONCLUSION
LCDK is a rare condition that is benign and does not necessiate surgery. However, if it causes clinical symptoms and hypertension, partial nephrectomy may be considered. While this condition may be morphologically identical to the ADPKD, it is not inherited and has no significant deterioration of renal function.[138] To the best of our knowledge, this is the first case of LCDK with clinical, laboratory (particularly, identification of source of elevated renin level) and radiological findings.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2013/3/1/33/116191
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- Localized cystic disease of the kidney: A rare entity. J Radiol Case Rep. 2012;6:29-35.
- [Google Scholar]
- Unilateral renal cystic disease: Report of an additional case of a rare disease. Port J Nephrol Hyper. 2011;25:1-3.
- [Google Scholar]
- Localized cystic disease of the kidney: Distinction from cystic neoplasms and hereditary polycystic diseases. Am J Surg Pathol. 2013;37:506-13.
- [Google Scholar]
- Localized cystic disease of the kidney: An unusual entity that can mimic a cystic neoplasm. Am J Kidney Dis. 2010;55:609-13.
- [Google Scholar]
- Localized cystic disease of the kidney: CT findings. Abdom Imaging. 2003;28:588-92.
- [Google Scholar]
- Unilateral and segmental localised polycystic kidney disease. J Clin Pathol. 1998;51:703-5.
- [Google Scholar]
- The pathogenesis of hypertension in autosomal dominant polycystic kidney disease. J Hypertens. 1997;15:925-33.
- [Google Scholar]




