
Translate this page into:
Neonatal Death Dwarfism in a Girl with Distinctive Bone Dysplasia Compatible with Grebe Chondrodysplasia: Analysis by CT Scan-based Phenotype
Address for correspondence: Dr. Ali Al Kaissi, Department of First Medical, Ludwig Boltzmann Institute of Osteology, Hanusch Hospital of WGKK and AUVA Trauma Centre Meidling, Hanusch Hospital, Heinrich Collin Str. 30 A-1140, Vienna, Austria. E-mail: Ali.alkaissi@oss.at
-
Received: ,
Accepted: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
We report on a female fetus noted to have severe malformative type of skeletal dysplasia on ultrasonography done at 35 weeks gestation. The girl died shortly after birth. Clinical examination showed a fetus with severe dwarfism, extensive long and short bones, and bone deficiencies associated with multiple dislocations. Computed tomography (CT) scan-based phenotype showed a complex constellation of malformations consistent with the diagnosis of Grebe syndrome. Parents being first cousins (consanguineous marriage) strongly suggests autosomal recessive pattern of inheritance. To our knowledge, this is the first report of neonatal death dwarfism of Grebe syndrome analyzed by CT scan-based phenotype.
Keywords
Computed tomography scan
Grebe dysplasia
long and short bones deficiency
neonatal death dwarfism

INTRODUCTION

The term neonatal death dwarfism indicates bone dysplasias. Such bone-related entities always have a fatal outcome. Skeletal dysplasias are genetically determined constitutional disorders of bone growth and development that result in radiographically demonstrable abnormalities of size, shape, density, or behavior of skeletal elements. Many are clinically apparent at birth, manifesting with either perinatal death or gross disproportions of head, body, and limbs, or some combination of these features. There are numerous forms of skeletal dysplasias which are almost lethal, such as thanatophoric dysplasia, achondrogenesis Type I and II, short rib syndromes, homozygous achondroplasia, atelosteogenesis, and boomerang dysplasia. If based on clinical grounds only, the diagnosis of these disorders is often uncertain or incorrect, whereas radiographic examination reveals fundamental diagnostic elements in a majority of cases.[12] Grebe chondrodysplasia is characterized by a distinctive phenotype. Major clinical features in patients with Grebe dysplasia are: Varying degrees of shortness of some or all fingers, knee abnormalities (dislocation and fixed flexion deformities), and foot deformities (mainly restricted ankle).[34] Grebe dysplasia is caused by mutations of CDMP1.[5] Parental consanguinity in our present case suggests autosomal recessive trait of inheritance.
No previous reports described lethality in patients with Grebe syndrome; nevertheless, we believe that lethality/mortality can occur in every disorder. Lack of reported cases cannot rule out this possibility. Besides, this is the first time CT phenotype was used. Previous reports describe the malformation complex via conventional radiographs. The latter cannot define/detect anatomical malformation complexes.
CASE REPORT
A stillborn girl fetus was born at 35 weeks gestation to first-degree cousins of a North African family. On ultrasonography, a single fetus with severe short and deformed limbs, severe skeletal dysplasia, and polyhydramnios was recognized. Parents chose to continue pregnancy. To relieve the mother from marked discomfort caused by polyhydramnios, 500 ml of amniotic fluid was removed by transabdominal amniocentesis and spontaneous labor occurred at 35 weeks gestation. Birth weight of the baby was 2.2 kg, length 38 cm, and head circumference 32 cm. The anterior and the posterior fontanelles were wide open. The head appeared large in comparison to the body. Parents permitted us to perform skeletal computed tomography (CT) scan but refused to allow autopsy. Clinical examination showed severe growth retardation and large head in comparison with the body (25th percentile). The main skeletal features were severe dwarfism associated with proximal, middle, and distal segments, and deficiency of the upper limbs associated with oligodactyly. Three-dimensional (3D) CT scan-based phenotype at gestational age of 35 weeks showed a large frontal area associated with bossing, depressed nasal bridge, hypertelorism, short snub nose, long philtrum, thin upper lip, microstomia, full cheeks, short thorax, a large bulging abdomen, oligodactyly, and genu recurvatum [Figure 1a]. Lateral view 3DCT scan-based phenotype showed low-set ears with folding defect and short neck. Severe shortening of the upper and lower limbs with flexion contracture and oligodactyly was also found [Figure 1b].
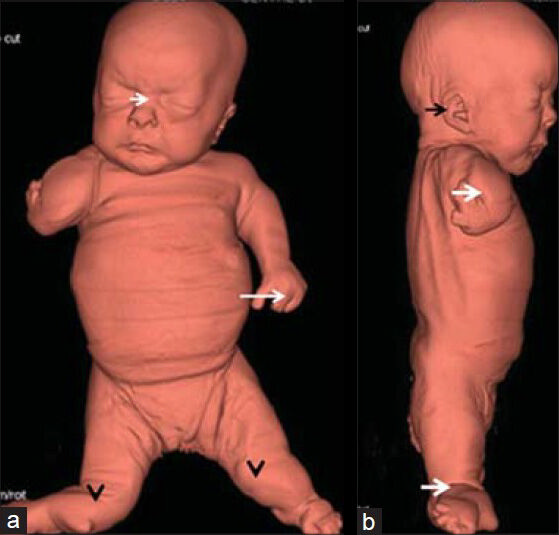
- Fetus at 35 weeks of gestation diagnosed with Grebe syndrome. 3DCT scan-based phenotype a) Anterior view shows a large frontal area associated with bossing, depressed nasal bridge (short white arrow), hypertelorism, short snub nose, long philtrum, thin upper lip, microstomia, full cheeks, short thorax, a large bulging abdomen, oligodactyly (long white arrow), and genu recurvatum (black arrowheads). b) Lateral view shows low-set ears with folding defect (black arrow), short neck, severe shortening of the upper and the lower limbs with flexion contracture and oligodactyly (white arrows).
Reconstruction skeletal CT scan of the fetus (anterior view) showed markedly opened anterior fontanel, defective development of the shoulder girdle, dysplastic lateral parts of the clavicles, aplasia of the glenoids, and marked tilting of the scapulae. Rudimentary humeri, aplasia of the radius on the right side and dysplastic radius and ulna of the left forearm associated with severe defective ossifications of the carpals were also found., though ossification to a certain extent of the distal phalanges of three digits was evident [Figure 2a]. Posterior view of the reconstruction skeletal CT scan showed markedly opened posterior fontanel, ill-defined cranio-cervical junction, and no anatomical landmark of the odontoid, associated with fusion of the posterior arches of C3-4 [Figure 2b]. Skeletal 3DCT scan confirmed large skull, massive ossification defects along the anterior fontanel, and severe gap along the coronal suture and another massive gap due to defective ossification along the metopic suture. The ischium appeared markedly dysplastic and was associated with double ossification of the capital femoral epiphyses leading to severe dysplasia of the pelvic girdle. Also found were aplasia of the left tibia and fibula, and a rudimentary right tibia [Figure 3]. Chromosomal analysis of the deceased neonate and her parents showed normal results.
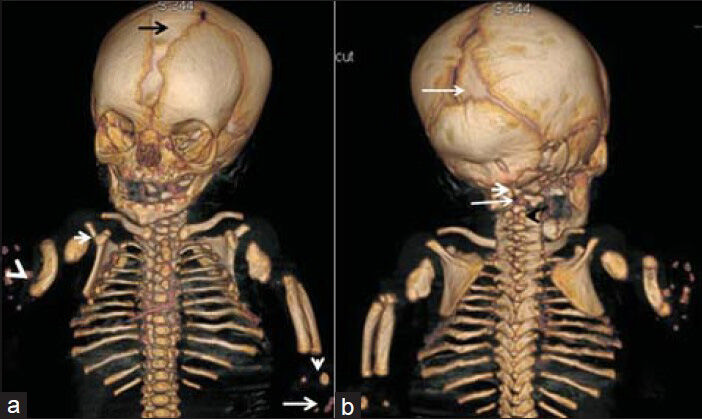
- Fetus at 35 weeks of gestation diagnosed with Grebe syndrome. Reconstruction skeletal CT scan a) Anterior view shows markedly opened anterior fontanel (black arrow), defective development of the shoulder girdle, dysplastic lateral parts of the clavicles, aplasia of the glenoids (white arrow), marked tilting of the scapulae, rudimentary humeri, aplasia of the radius on the right side (radial club hand and dislocation) (white arrowhead), and dysplastic radius and ulna of the left forearm associated with severe defective ossification of the carpals (vertical white arrow), though ossification of the distal phalanges of three digits is evident to a certain extent (long white arrow). b) Posterior view shows markedly opened posterior fontanel (long white arrow), ill-defined cranio-cervical junction (short white arrow), and no anatomical landmark of the odontoid (long white arrow), associated with fusion of the posterior arches of C3-C4 (black arrowhead).
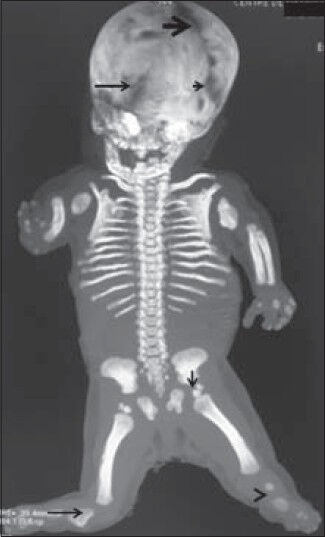
- Fetus at 35 weeks of gestation diagnosed with Grebe syndrome. Skeletal 3DCT scan confirms large skull and massive ossification defects along the anterior fontanel (black thick arrow), severe gap along the coronal suture (short black arrow) and another massive gap (defective ossification) along the metopic suture (long black arrow). The ischium appears markedly dysplastic and is associated with double ossification of the capital femoral epiphyses (vertical black arrow) (severe dysplasia of the pelvic girdle), aplasia of the left tibia and fibula (black arrowhead) and presence of a rudimentary right tibia (long black arrow).
DISCUSSION
The phenotypic features in our patient diagnosed with Grebe Chondrodysplasia were distinctive and different from all previously published forms of neonatal dwarfism. Seventeen cases of neonatal death dwarfism have been reported. They include Majewski syndrome, Meckel syndrome, homozygous achondroplasia, rhizomelic type of punctate epiphyseal dysplasia, cloverleaf skull syndrome, lethal form of osteogenesis imperfecta, campomelic dwarfism, and achondrogenesis syndromes. Seven cases of rare “neonatal death dwarfism” observed in three major Children's Hospitals in Australia are reported. These include diastrophic dwarfism, achondrogenesis II, lethal form of hypophosphatasia, homozygous achondroplasia, minor form of asphyxiating thoracic dystrophy, and unclassified lethal bone dysplasia.[126] None of the above-mentioned clinical entities seem similar to the form seen in this neonate.
Severe micromelia is a frequent abnormality in patients with atelosteogenesis Type II (a lethal chondrodysplasia). Skeletal abnormalities such as coronal clefts of the vertebral bodies, metaphyseal and epiphyseal abnormalities, talipes equinovarus, and abducted thumbs and toes are characteristic presentations.[7] The overall skeletal abnormalities are totally different from those of our present case. Acromesomelic dysplasia is a short stature syndrome first reported by Maroteaux et al. It shows uniform shortening of the digits and hypoplastic/dysplastic vertebral bodies. The head is relatively large with frontal bossing. The radial head might be dislocated.[8]
Other forms of mesomelic shortening such as Du pan syndrome are characterized by acromesomelic dysplasia associated with mesomelic shortening of the extremities, very short fingers, and nubbin-like toes. Radiographic findings include absent fibulae, hypoplastic or dysplastic forearms, dysplasia or absence of the middle and proximal phalanges, and carpal fusion in some patients.[9]
In 1981, Langer et al., described Grebe syndrome as a clinical entity akin to Du Pan and Hunter Thompson syndromes. The phenotypes in these syndromes are distinctive and regarded as different entities from that seen in our neonate. Patients with Grebe dysplasia do manifest severe peripheral skeletal abnormalities. These patients are much shorter as a result of severe involvement of the long bone.[10]
CONCLUSION
Distinct features in our present case diagnosed as Grebe Chondrodysplasia include short-limbed dwarfism with a relatively large head and unusual facial features of hypertelorism, depressed nasal bridge, short-snub nose, large and low-set ears with folding defects, and full cheeks. Hypermobile joints and dislocated elbows, wrists, tibia, and ankle joints were present. Oligodactyly, shoulder girdle defects, and radial club hand were the other features. Interestingly, by means of 3D-CT scan, we were able to detect a constellation of cranial malformations such as severe defective ossification of the fontanelles, sutures, and the ill-defined cranio-cervical junction, as well as fusion of the posterior arches of C3-C4. However, no genetic test was done in the present case.
ACKNOWLEDGMENT
We wish to thank Professor Andrea Superti-Furga Leenaards, Chairman of Pediatrics, Rare Bone Diseases and Skeletal Dysplasia, University of Lausanne for confirming the diagnosis in this neonate.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2014/4/1/53/141939
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- Die Achondrogenesis. ein einfach rezessives Erbmerkmal. Folia Hered Path. 1952;23:2-8.
- [Google Scholar]
- Grebe syndrome: Clinical and radiographic findings in affected individuals and heterozygous carriers. Am J Med Genet. 1988;75:523-9.
- [Google Scholar]
- Du Pan syndrome phenotype caused by heterozygous pathogenic mutations in CDMP1 gene. Am J Med Genet A. 2005;138:379-83.
- [Google Scholar]
- London Dysmorphology Database, London Medical Databases. Oxford: Oxford University Press; 2009.
- [Google Scholar]
- Absence congenitale du perone sans deformation du tibia: Curieuses deformations congenitales des mains. Rev Orthop. 1924;11:227-34.
- [Google Scholar]
- A severe autosomal recessive acromesomelic dysplasia, the Hunter-Thompson type, and comparison with the Grebe type. Hum Genet. 1989;81:323-8.
- [Google Scholar]




