
Translate this page into:
Vaginal Ewing Sarcoma: An Uncommon Clinical Entity in Pediatric Patients
Address for correspondence: Dr. A Luana Stanescu, Seattle Children's Hospital, 4800 Sand Point Way NE, Seattle, WA 98105, USA. E-mail: stanescu@u.washington.edu
-
Received: ,
Accepted: ,
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Ewing sarcoma, including classical Ewing sarcoma of the bone and primitive neuroectodermal tumors arising in bone or extraosseous primary sites, is a highly aggressive childhood neoplasm. We present two cases of Ewing sarcoma arising from the vagina in young girls. Previously reported cases in literature focused on their pathologic rather than radiographic features. We describe the spectrum of multimodality imaging appearances of Ewing sarcoma at this unusual primary site. Awareness of vaginal Ewing tumors may facilitate prompt diagnosis and lead to a different surgical approach than the more commonly encountered vaginal rhabdomyosarcoma.
Keywords
Ewing sarcoma
pediatric
vaginal


Introduction
The World Health Organization currently uses the term Ewing sarcoma to include classical Ewing of the bone as well as extraosseous Ewing, Askin tumor of the thoracic wall, and primitive peripheral neuroectodermal tumors.[1] Because Ewing sarcoma has a common biology and treatment regardless of the primary site or degree of cellular differentiation, distinctions based on location and histologic variations are no longer utilized for classification or therapy. Virtually, all Ewing sarcomas share a common chromosomal translocation of the long arm of chromosome 11 and 22. The translocation of the EWSR1 gene on to chromosome 22p12 next to the FLI1 gene usually results in upregulation of insulin-like growth factor 1 which plays a role in cellular proliferation.[2] The common translocation and a strong membranous expression of CD99 unify the diagnosis of classical and extraosseous Ewing sarcoma, supporting the use of the new nomenclature.
Ewing sarcoma is the second most common malignancy of bone with an incidence of approximately 200 cases per year in the United States in children under 20.[3] A recent study by Applebaum et al. found that extraskeletal Ewing sarcoma represents approximately 31% of all cases of Ewing sarcoma.[4]
Extraosseous Ewing sarcoma originating in the vagina is rare, with <10 cases reported to date in the literature.[5] We present two cases occurring in teenagers, one of them being the youngest case reported so far. These highly aggressive tumors require a more intensive treatment regimen compared to other vaginal masses such as rhabdomyosarcoma. While the imaging findings are nonspecific, awareness of this clinical entity can facilitate early diagnosis and appropriate treatment, which may improve prognosis.
Case Reports
Case 1
A 12-year-old female began menarche with heavy bleeding and menorrhagia. Use of oral contraceptives resulted in a notable decrease in symptomatology. Subsequent acute onset of urinary retention prompted an Emergency Department (ED) visit 3 months later. Pelvic ultrasound was performed, demonstrating a large solid, heterogeneous mass, located posterior to the bladder, presumably vaginal [Figure 1a] which was further characterized by a contrast-enhanced magnetic resonance (MR) examination [Figure 1b–f], obtained after placement of a Foley catheter. On MR imaging (MRI), the lesion was confirmed to arise from the vagina. The patient was transferred to our hospital for further management with a presumptive diagnosis of rhabdomyosarcoma. Biopsy was performed. Subsequent staging 18Fluorine fluorodeoxyglucose positron emission tomography/computed tomography (18F FDG-PET or PET/CT) [Figure 1g] was obtained, identifying multiple pulmonary metastases as well as a metastatic lesion at the T11 vertebral body. Histology [Figure 1h] demonstrated a round blue cell tumor with scattered areas of necrosis and moderate mitotic activity.
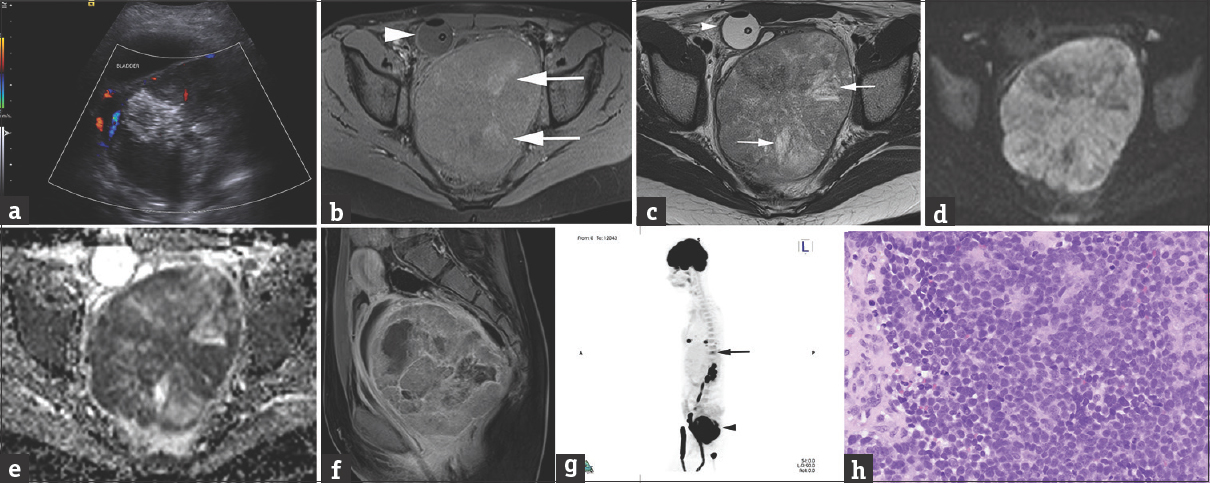
- A 12-year-old female presented with acute onset of urinary retention. (a) Transverse, color flow ultrasound image through the pelvis demonstrates a large, heterogeneous mass posterior to the bladder measuring 10.1 cm × 8.8 cm × 12.4 cm (anteroposterior × transverse × craniocaudal), with minimal vascularity, and large hyperechoic foci, some of which shadow, suggestive of calcifications. (b) Axial T1 fat-sat precontrast MRI with a Foley decompressing the bladder (arrowhead) better depicts the large heterogeneous pelvic mass, containing areas of high T1 signal (arrows) corresponding to calcifications present on low-dose noncontrast CT for attenuation correction performed as part of the 18F FDG-PET (not shown). (c) Axial T2 image at a similar level as b shows areas of high T2 signal representing cystic necrosis. (d and e) Axial diffusion and apparent diffusion coefficient map images show extensive restrictive diffusion. (f) Sagittal contrast-enhanced T1 MRI of the pelvis confirms that the large, heterogeneously enhancing mass with areas of necrosis arises from the posterior wall of the vagina, compresses the bladder and the rectum and displaces the uterus superiorly. (g) Sagittal three-dimensional maximum intensity projection 18F FDG-PET/CT demonstrates heterogeneous hypermetabolic uptake in the pelvic mass (arrowhead) as well as additional hypermetabolic foci in the lung bases and at the T11 vertebral body (arrow), consistent with metastatic lesions. (h) Histologic examination shows sheets of small round blue cells with inconspicuous nucleoli (H and E, ×400). The cells are focally arranged in primitive rosettes (top center).
The tumor cells had diffuse membranous CD99 expression by immunohistochemistry, and the EWSR1 gene rearrangement was detected by fluorescent in situ hybridization (FISH), consistent with a diagnosis of Ewing sarcoma. Bone marrow aspiration and biopsy were negative for marrow metastases. Chemotherapy with vincristine, cyclophosphamide, and doxorubicin alternating with ifosfamide and etoposide was instituted.[6] After completion of chemotherapy, the patient developed progressive metastatic lung disease and a left parietal metastatic lesion, being currently managed with palliative treatment.
Case 2
A 15-year-old female with progressive abdominal fullness and discomfort, profuse menstrual bleeding, and malodorous vaginal discharge for 3 months presented to the ED with a lobulated, vascular mass protruding from the vaginal introitus.
Pelvic ultrasound demonstrated a 4 cm vaginal mass [Figure 2a], further characterized by MRI [Figure 2b and c] as arising from the left vaginal sidewall with possible extension into the adjacent levator ani musculature.18F FDG-PET/CT showed radiotracer uptake in the primary pelvic tumor [Figure 2d]. No metastatic disease identified on bone scintigraphy,18F FDG-PET/CT or bone marrow aspiration, and biopsy. Biopsy specimens demonstrated a round blue cell tumor with abundant mitotic figures and Horner–Wright type rosettes [Figure 2e]. Immunohistochemical stain results included diffuse membrane expression of CD99. Stains for rhabdomyosarcoma (desmin, myogenin, and MyoD1) were negative. FISH demonstrated EWSR1 rearrangement without SYT rearrangement, confirming extraosseous Ewing sarcoma.
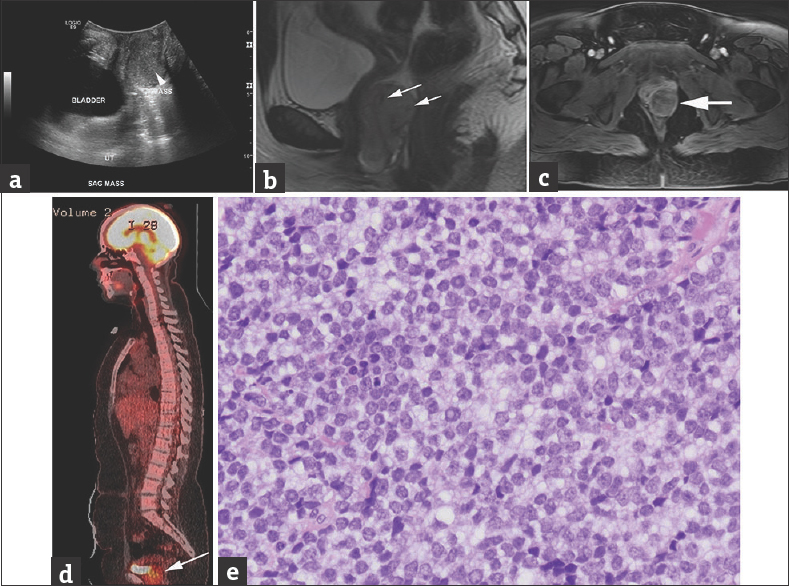
- A 15-year-old female presented with progressive abdominal discomfort, profuse menstrual bleeding, and vaginal discharge. (a) Grayscale sagittal ultrasound image through the pelvis obtained at initial presentation demonstrate a slightly heterogeneous, predominantly isoechoic, 3.6 cm × 3.6 cm × 5.9 cm (anteroposterior x transverse x craniocaudal), vaginal mass (arrowhead on a). (b and c) Sagittal T2 (b) and axial postcontrast T1 MRI (c) show the lesion arising from the left vaginal sidewall demonstrating intermediate to high signal on T2-weighted images with few serpentine flow voids (arrows on b) and intermediate to low T1 signal, with heterogeneous contrast enhancement on (c). (d) Sagittal fused 18F FDG-PET/CT demonstrates increased FDG uptake in the mass (arrow) posterior to the bladder. (e) Histologic examination shows sheets of small round blue cells with inconspicuous nucleoli and a suggestion of rosette formation (H and E, ×400 magnification). Pathology image courtesy of Dr. Aashiyana Koreishi, Puget Sound Institute of Pathology - Franciscan Health System.
The patient started chemotherapy with vincristine, doxorubicin, and cyclophosphamide alternating with ifosfamide and etoposide. Following 12 weeks of neoadjuvant chemotherapy, a complete surgical resection of the posterior vaginal mass with preservation of the vagina was performed. Since the resected tumor was >90% necrotic without extension to the surgical margins, no adjuvant radiation therapy was given. The patient had no evidence of recurrent disease 12 months after therapy completion.
Discussion
Extraskeletal Ewing tumors are usually rapidly growing deep soft tissue masses, often presenting with pain and/or a large mass (average 5–10 cm at presentation), similar to our cases. Time to diagnosis is one of the longest as compared to other pediatric tumors; however, it is not associated with worsened prognosis.[7] The most frequent sites of origin in decreasing order of the prevalence include the paravertebral region, lower extremity, chest wall, retroperitoneum, pelvis, and hip.[7] The lung is the most common site of metastatic spread.[8]
Pathologically, these tumors are often well demarcated with early involvement of adjacent soft tissues occurring in many cases. Histology shows sheets of small round blue cells sometimes intermixed in a fibrous stroma. Necrosis and hemorrhage are not uncommon.
The imaging appearance of extraosseous Ewing sarcoma is usually that of a hypervascular soft tissue mass with areas of necrosis or hemorrhage, which may abut adjacent bone but does not involve the marrow space. Calcifications are present in up to 25% of cases.[3] The proposed diagnostic imaging criteria for extraskeletal Ewing sarcoma rely on the absence of adjacent bony invasion on MRI with no abnormal uptake on delayed imaging technetium-99m methylene diphosphonate (99m Tc-MDP) bone scan.[3]
CT usually reveals an isodense mass which can have an indistinct or infiltrating margin with some degree of enhancement.[8] Ultrasonographic features include a hypoechoic lesion, which may be heterogeneous due to necrosis, hemorrhage, or calcification. Similar heterogeneity is seen on MRI with the lesions being T2 isointense to hyperintense, T1 isointense to muscle, with heterogeneous contrast enhancement. Serpentine vessels with flow voids are often seen traversing the lesion, which may be surrounded by a pseudocapsule. The lesions are typically hypermetabolic on18F FDG-PET/CT although non-FDG-avid lesions have been described.[38] Bone scintigraphy usually demonstrates increased uptake on blood flow and blood pool images, reflecting the hypervascular nature of these lesions, without significant increased uptake on delayed images.[39]
Both our cases demonstrate a heterogeneous appearance on ultrasound and MRI, more conspicuous in the first case where a larger mass with extensive calcifications and area of necrosis was present. Minimal necrosis and no definite calcifications characterized the second case, possibly due to the smaller size of the mass. Neither of these lesions demonstrated significant Doppler color flow although a few serpentine flow voids were noted on MR in the second case. Both lesions showed restricted diffusion and heterogeneous contrast-enhancement on MRI. Both primary and metastatic lesions were found to be FDG-avid. The second case did not show definite radiotracer uptake within the mass on blood pool or delayed images on99m Tc-MDP whole-body scintigraphy study. This examination was not performed for the first patient.
The outcome for Ewing sarcoma is primarily dependent on the presence of distant metastases. In the absence of detectable metastases, 70% of children and adolescents are cured with a combination of intensive, multiagent chemotherapy with surgery and/or radiotherapy. In contrast, <30% of children and adolescents with overt metastases can be cured with current therapy.[10]
Although the imaging features of extraskeletal Ewing sarcoma are not specific, they suggest an aggressive, rapidly proliferating mass, which should prompt clinicians to obtain early biopsy confirming pathologic diagnosis. The current consensus for treatment of these lesions includes chemotherapy and delayed surgical resection when feasible, supplemented with radiotherapy if indicated.[5] Initial misdiagnosis can lead to incomplete treatment with recurrent disease. The only case reported treated with partial resection and radiotherapy, misdiagnosed initially as an undifferentiated carcinoma, presented with a cranial metastasis 1 year later.[11]
Since imaging is not definitive, a well-structured differential diagnosis for a pediatric or adolescent patient with an aggressive pelvic (extraovarian) lesion should include rhabdomyosarcoma, extraskeletal Ewing sarcoma, synovial sarcoma, malignant melanoma, and less frequently carcinoma.[51213] While biopsy is a key in confirming the diagnosis, imaging remains critical in defining the extent of disease, in evaluating response to treatment, and in the detection of recurrent/metastatic disease.18F FDG-PET can play an important role in guiding the biopsy of these heterogeneous, often partially necrotic, vascular tumors.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2017/7/1/17/205176
References
- Ewing sarcoma. In: WHO Classification of Tumours of Soft Tissue and Bone. Lyon: IARC Press; 2013. p. :305-9.
- [Google Scholar]
- Preliminary efficacy of the anti-insulin-like growth factor type 1 receptor antibody figitumumab in patients with refractory Ewing sarcoma. J Clin Oncol. 2011;29:4534-40.
- [Google Scholar]
- From the radiologic pathology archives: Ewing sarcoma family of tumors: Radiologic-pathologic correlation. Radiographics. 2013;33:803-31.
- [Google Scholar]
- Clinical features and outcomes in patients with extraskeletal Ewing sarcoma. Cancer. 2011;117:3027-32.
- [Google Scholar]
- Primary vaginal Ewing sarcoma: Case report and review of the literature. Int J Surg Pathol. 2012;20:305-10.
- [Google Scholar]
- Randomized controlled trial of interval-compressed chemotherapy for the treatment of localized Ewing sarcoma: A report from the Children's Oncology Group. J Clin Oncol. 2012;30:4148-54.
- [Google Scholar]
- Time to diagnosis of Ewing tumors in children and adolescents is not associated with metastasis or survival: A prospective multicenter study of 436 patients. J Clin Oncol. 2014;32:1935-40.
- [Google Scholar]
- Multimodality imaging features, metastatic pattern and clinical outcome in adult extraskeletal Ewing sarcoma: Experience in 26 patients. Br J Radiol. 2014;87:20140123.
- [Google Scholar]
- Appearance of extraosseous pelvic Ewing sarcoma on triphasic bone scan. Clin Nucl Med. 2014;39:406-8.
- [Google Scholar]
- Ewing sarcoma. In: Pizzo PA, Poplack DG, eds. Principles and Practice of Pediatric Oncology (7th ed). Philadelphia: LWW; 2015. p. :854-75.
- [Google Scholar]
- Primary vaginal extraosseous Ewing sarcoma/primitive neuroectodermal tumor with cranial metastasis. J Chin Med Assoc. 2009;72:332-5.
- [Google Scholar]
- Ewing family of tumours involving the vulva and vagina: Report of a series of four cases. J Clin Pathol. 2007;60:674-80.
- [Google Scholar]
- Primary vaginal Ewing's sarcoma or primitive neuroectodermal tumor in a 17-year-old woman: A case report. J Med Case Rep. 2010;4:88.
- [Google Scholar]




