
Translate this page into:
Primary Pulmonary Chondrosarcoma: A Case Report and Literature Review

-
Received: ,
Accepted: ,
How to cite this article: Yasin JT, Daghlas SA, Hamid A, Gaballah AH. Primary Pulmonary Chondrosarcoma: A Case Report and Literature Review. J Clin Imaging Sci 2020;10:3.
Abstract
Chondrosarcomas are tumors consisting of osseous or cartilaginous stroma. They are not an uncommon pathology; however, primary pulmonary chondrosarcomas arising in lung parenchyma are extremely rare, with few cases published in literature. Herein, we present a case with biopsy-proven primary pulmonary chondrosarcoma after exclusion of primary origin elsewhere. In the case presented in this report, we demonstrate the clinical presentations, pulmonary function tests, and the radiological findings of this rare tumor in a young male patient. Further, we present a brief review of existing literature for patients with similar pathology.
Keywords
Primary
Pulmonary
Chondrosarcoma

INTRODUCTION
Chondrosarcomas of primary lung parenchymal origin are an extremely rare pathology with most descriptions coming from a series of case reports and small case series. These tumors can present as centrally located, parenchymal masses. Diagnosis requires extensive workup to exclude alternative etiology and to confirm diagnosis. To date, 28 cases, not including the current study, of this pathology have been reported. Herein, we present a case with biopsy-proven primary pulmonary chondrosarcoma and a brief literature review of similar cases.
CASE REPORT
A 23-year-old Caucasian male presented to a cardiothoracic surgery clinic for follow-up of a lung mass. The mass was identified on chest X-ray and computed tomography (CT) chest studies that were performed for the evaluation of a non-productive cough, fever, and 10 pounds weight loss.
On physical examination, the patient exhibited mild expiratory wheezing bilaterally. Pertinent laboratory results showed hypercalcemia. Pulmonary function tests were indicative of an obstructive lung disease; however, a bronchoscopy examination was unremarkable. Chest X-ray and contrast-enhanced CT were performed, revealing the presence of a large mass with several intrinsic calcifications in the upper lobe of the right lung along the paramediastinum [Figures 1 and 2]. An ultrasound-guided procedure was conducted to localize the lesion and biopsy the surrounding lymph nodes and was indicative of low-grade cartilaginous neoplasm, presumed to be either chondroma or teratoma.
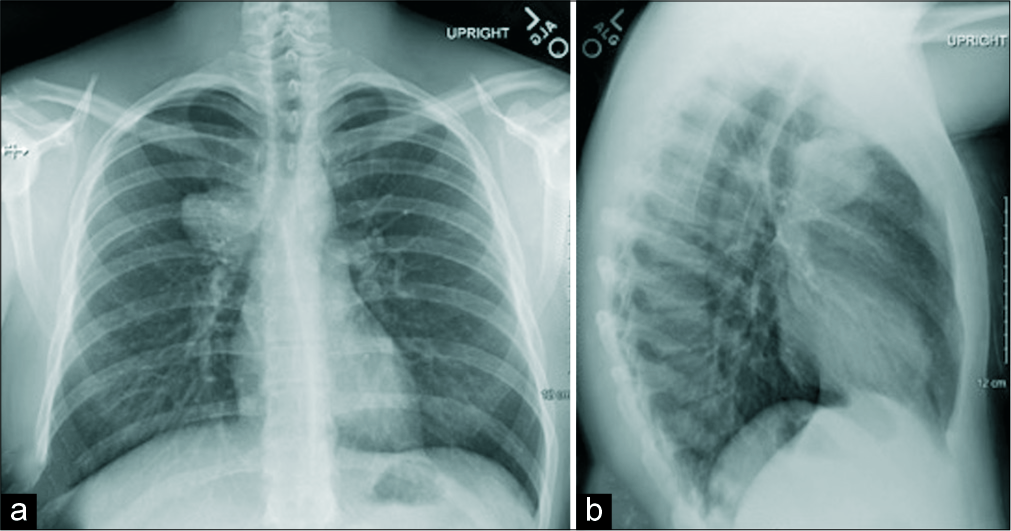
- Chest radiograph frontal (a) and lateral (b) views of a 23-year-old male demonstrate rounded mass-like opacity (arrows) along the superior right paramediastinal border in the right upper lobe.
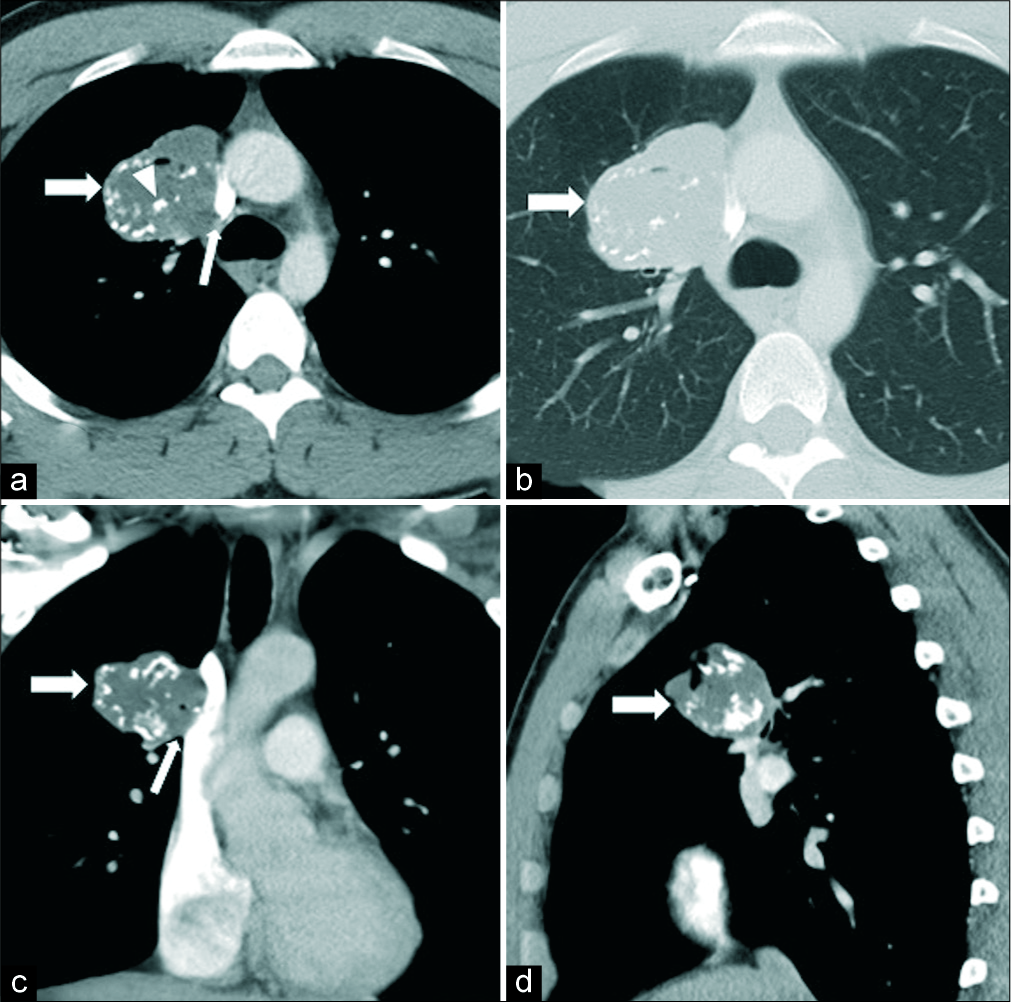
- Contrast-enhanced computed tomography chest in axial (a and b), coronal (c), and sagittal (d) views of a 23-year-old male revealed a large soft tissue mass in the right upper lobe (thick arrows) along the right paramediastinal border abutting the adjacent mediastinal vascular structures (thin arrows), with multiple intrinsic calcifications (arrowhead). Surgery resection of the mass was performed and the lesion proved to be a low-grade primary lung chondrosarcoma.
An exploratory right video-assisted thoracoscopic surgery was performed as primary treatment. The operation revealed an intraparenchymal central mass in the right upper lobe [Figure 3] adherent to the mediastinum adjusted to the main pulmonary artery, thus necessitating a lobectomy. Postsurgical CT scan of the chest and bone scan using Tc-99m diphosphate revealed no residual tumor or secondary lesions, respectively.
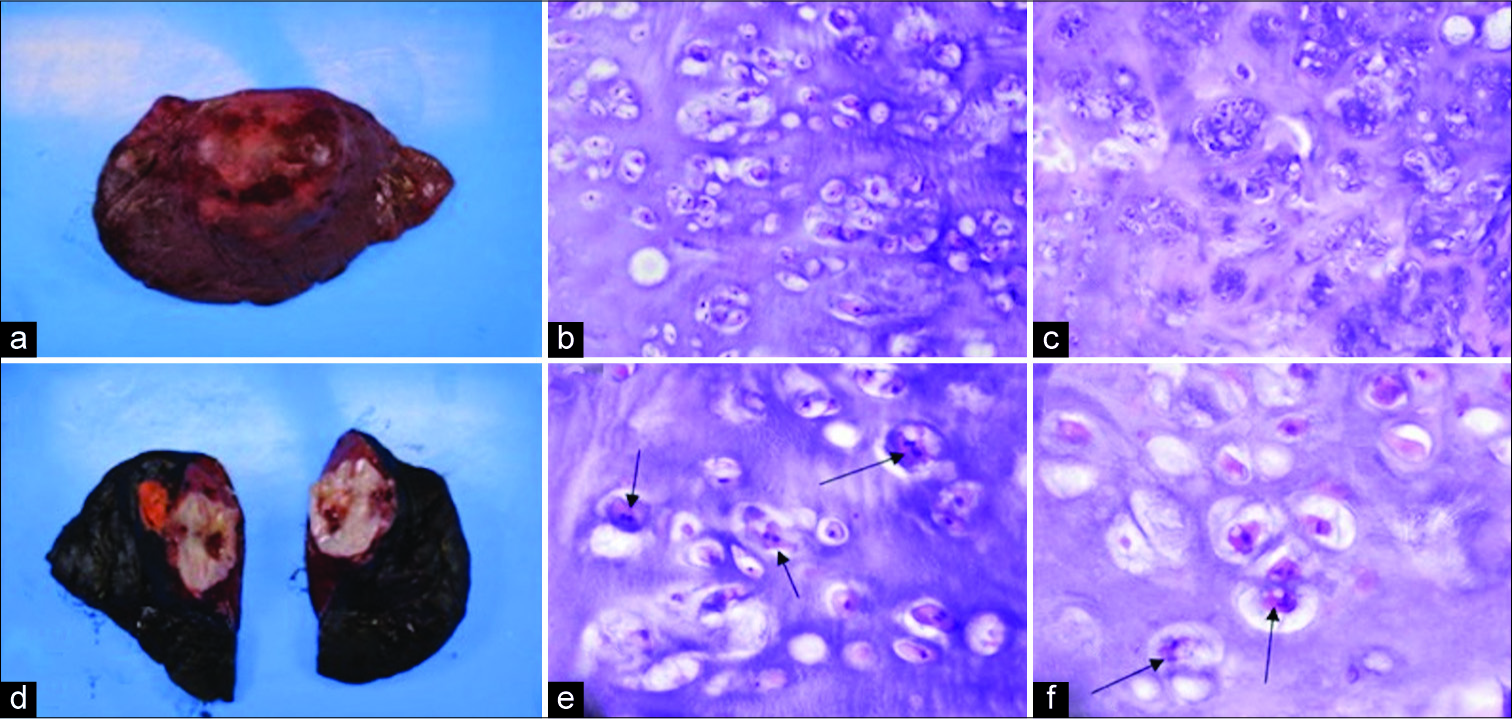
- Resected 5 cm primary chondrosarcoma from a 23-year-old male from the upper lobe of right lung through exploratory video-assisted thoracoscopic surgery, (a and b) gross appearance of resected mass, (c-f) histopathological findings of primary pulmonary chondrosarcoma with H and E stain, arrows point toward binuclear chondrocytes with nuclear crowding, which suggest the presence of a low-grade chondrosarcoma.
A histopathological review revealed a well-demarcated lobulated, rubbery, and cartilaginous tumor with a focal necrotic appearance and calcifications. The lesion had stromal hyalinization and prominent stromal vessels with mild atypia. Immunostains for pankeratin, PLAG1, and HMGA2 were negative. The presence of cellular crowding and binuclear chondrocytes [Figure 3] supported the final interpretation of the lesion as a low-grade (Grade 1) pulmonary chondrosarcoma.
DISCUSSION
Chondrosarcomas of primary lung parenchymal origin are extremely infrequent with most descriptions of the pathology coming almost exclusively from case reports, as depicted in Table 1. A literature search of PubMed using the query [(pulmonary OR lung) AND chondrosarcoma AND primary] yielded 14 studies with full-text available or sufficient information available in the abstract. Ten additional studies were found through a bibliographic review. Cases that did not originate from lung parenchymal tissue were excluded from the study. Such excluded cases include pulmonary artery, costal, and bronchial chondrosarcomas.
| Study primary author, year | Age/sex | Presentation | Location | Type | Treatment | Outcome |
|---|---|---|---|---|---|---|
| Balanzá, 2016[5] | 69/M | Hemoptysis | RLL | Myxoid | oracotomy | Complete surgical resection. No evidence of recurrence for 6 months of follow-up |
| Jiang, 2016[4] | 59/M | Hemoptysis, cough, dyspnea | RUL | Myxoid | Surgical removal, ifosfamide, doxorubicin | Distal metastases, Ongoing treatment |
| Endicott, 2015[6] | 69/M | Acute-onset dyspnea | LUL | Myxoid | Sublobar resection | Complete surgical resection. No evidence of recurrence for 6 months of follow-up |
| Wang, 2014[1] | 52/F | Intermittent cough, dyspnea | LUL | Dedifferentiated | oracotomy | Patient is alive after 36 months of follow-up |
| Mei, 2013[7] | 20/F | Non-productive cough, chest pain | LLL | Mesenchymal | Pneumonectomy | Lost to follow-up |
| Kalhor, 2011[2] | 1. 69/F 2. 69/M 3. 53/M 4. 51/F |
1. NA 2. NA 3. NA 4. NA |
1. NA 2. NA 3. NA 4. NA |
1. Hyaline 2. Myxoid 3. Myxoid 4. Hyaline |
1. NA 2. NA 3. NA 4. NA |
1. NA 2. Decreased, 45 days post-operative 3. NA 4. Patient is alive after 36 months of follow-up |
| Li, 2011[8] | 51/F | Severe anemia | LUL | Myxoid | oracotomy, left upper lobectomy with systemic lymph node dissection | Smooth recovery, discharged in good condition without anemia after 2 weeks, follow-up every 3 months, no abnormalities32 months post-operative |
| Boueiz, 2009[9] | 57/F | Exertional dyspnea | RLL, posterior mediastinum | Myxoid | oracotomy, complete debulking, pleurodesis | Uneventful post-operative course, no recurrence of pleural effusion, declined adjuvant chemotherapy, and lost to follow-up |
| Shah, 2007[10] | 60/M | Dry cough | LLL | Hyaline | oracotomy, recurrence/metastasis treated with radiotherapy, ifosfamide, doxorubicin, surgical resection | Full recovery after treatment of distal metastases. |
| Steurer, 2007[11] | 49/M | Chest pain, fatigue, weight loss | LUL | Dedifferentiated | Doxorubicin, ifosfamide, surgical resection | No evidence of further tumor lesions after 30 months of follow-up |
| Ichimura, 2005[12] | 35/M | Incidental | LLL | Myxoid | VATS | Patient is alive after 72 months of follow-up |
| Huang, 2002[13] | 40/M | Incidental | LUL | Mesenchymal | oracotomy | No evidence of recurrence/metastasis after 12 months of follow-up |
| Parker, 1996[14] | 25/F | Abdominal pain | LUL | Hyaline | Surgical excision | Patient is alive after 14 months of follow-up |
| Hayashi, 1993[15] | 73/M | Incidental | RML | Hyaline | Surgical resection | Metastases to skull, kidneys |
| Kurotaki, 1992[16] | 45/F | NA | RLL | Mesenchymal | Lobectomy | Patient is alive after 12 months of follow-up |
| Stanfield, 1991[17] | 78/M | Dull pain in the left chest and shoulder | LUL | Mesenchymal | oracotomy, resection of LUL, 2ndand 3rdribs, and adjacent soft tissue | Normal post-operative recovery with a significant reduction in cancer-related pain,tumor identified in a subclavian nodule of the chest wall adjacent to surgical incision 2months later |
| Watanabe, 1990[18] | 67/M | Fever, cough | RLL | Myxoid/Hyaline | Lobectomy | No signs of recurrence after 44 months |
| Jazy, 1984[19] | 1. 61/F 2. 70/M |
1. Non-productive cough, right-sided pleuritic chest pain. 2. Left-sided chest pain, let scapular pain, left arm pain, left Horner syndrome, signs of cord compression with paraparesis and altered sensorium |
1. RLL, lower posterior mediastinum. 2. LUL, extending into epidural space at the level of T2 |
1. NA 2. NA |
1. Exploratory thoracotomy, radiation, incomplete resection, cyclophosphamide, doxorubicin 2. Decompressive laminectomy ×2, radiation, physical rehabilitation |
1. Recurrence 5 months post-operative. Died of cardiac causes 4 months later 2. Progressive debilitation, death within 7 months of presentation. |
| Sun, 1982[20] | 31/F | Wheezing, cough | RLL | Myxoid/Hyaline | Pneumectomy, radiotherapy | Deceased |
| Morgan, 1972[21] | 23/F | Incidental | RLL | Myxoid | oracotomy, local excision | Discharged on post-operative day 9. Doing very well on follow-up 15 months post-operative. |
| Rees, 1970[22] | 64/M | Cough with purulent sputum | LUL | Hyaline | Lobectomy | Patient is alive after 48 months of follow-up |
| Smith, 1960[23] | 53/F | Chronic fatigue, frontal headache, palpitations, nocturia, ankle swelling | Right lung | NA | NA | NA |
| Lowell, 1949[24] | 41/F | Fatigue, anorexia, lower extremity edema, weight loss, dyspnea, tachycardia, palpitations | LLL | Mesenchymal | No treatment, patient decided to forgo thoracotomy | Patient died of expanding the pneumothorax |
| Greenspan, 1933[25] | 35/F | Chest pain | LUL | NA | None (diagnosed at autopsy) | Deceased |
M: Male, F: Female, NA: Not available, RUL: Right upper lobe, RML: Right middle lobe, RLL: Right lower lobe, LUL: Left upper lobe, LLL: Left lower lobe, VATS: Video-assisted thoracoscopic surgery
The incidence of primary pulmonary chondrosarcoma is difficult to estimate as this pathology is frequently misdiagnosed, requiring an extensive clinical history and diagnostic workup to exclude alternative primary origin; thus, it may be underreported in the literature. Patients with pulmonary chondrosarcomas frequently present with symptomatic pulmonary obstruction, dyspnea, and chest pain. The tumors are often centrally located and present with sharp borders. CT scans that demonstrate a well-demarcated, centrally located mass with calcifications may be indicative of pulmonary chondrosarcoma and this pathology should be included in the differential diagnosis of patients with similar presentations to ours [Figures 1 and 2].
The presence of calcifications is disputed as a case report claimed calcifications are classically present,[1] whereas other case reports indicate an absence of calcifications.[2] Histologically, pulmonary chondrosarcomas are characterized by ill-defined lobules containing differentiated hyaline cartilage with plump nuclei, binucleation, hyperchromasia, and coarse chromatin.[2] The presence of basophilic myxoid stroma with focal areas of hyaline cartilage may also be seen.[2] Immunohistochemical studies of the tumors are negative for keratin, CD31, and desmin and are S-100 positive.
To diagnose a primary pulmonary chondrosarcoma, a detailed clinical history and appropriate imaging are essential to exclude the following criteria: Prior history of skeletal neoplasms, a thoracic cage neoplasm, limb amputation, and teratoma.[2] Furthermore, the presence of the Carney triad (pulmonary chondroma along with gastrointestinal stromal tumor and extra-adrenal paragangliomas) should be excluded.[3]
Regarding treatment, complete surgical resection of the tumor is recommended as malignant cartilage may be observed with mixed epithelial/mesenchymal neoplasms.[2] Complete resection of the tumor may also prevent misdiagnosis based on improper biopsy specimen as teratomas may resemble pulmonary chondrosarcoma from biopsy location inaccuracy.[4]
A comprehensive review of existing literature on this tumor reveals that these tumors, when treated early, generally respond well to surgical treatment. These tumors have been known to metastasize to distal sites, such as the skull and kidneys, where adjuvant chemotherapy may be more beneficial. Further, investigation and study of the radiological findings in cases of primary pulmonary chondrosarcomas and reported outcomes are needed to establish reliable diagnostic criteria and treatment recommendations.
CONCLUSION
Primary pulmonary chondrosarcomas are a rare form of chondrosarcomas as seen in the dearth of cases reported in literature. We report a case which was prompted by the identification of a lung mass when the patient came in for chest X-ray and chest CT for fever, non-productive cough, and weight loss. Diagnostic workup of exclusion and histological analysis confirmed its origin from lung parenchymal tissue. It is necessary to pursue further study of radiological findings of primary pulmonary chondrosarcomas to develop guidelines and criteria to reliably establish its diagnosis.
Declaration of patient consent
Patient’s consent not required as patients identity is not disclosed or compromised.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- Primary chondrosarcoma presenting as an intrathoracic mass: A report of three cases. Oncol Lett. 2014;8:1151-4.
- [CrossRef] [PubMed] [Google Scholar]
- Primary pulmonary chondrosarcomas: A clinicopathologic study of 4 cases. Hum Pathol. 2011;42:1629-34.
- [CrossRef] [PubMed] [Google Scholar]
- The triad of gastric leiomyosarcoma, functioning extra-adrenal paraganglioma and pulmonary chondroma. N Engl J Med. 1977;296:1517-8.
- [CrossRef] [PubMed] [Google Scholar]
- Primary pulmonary chondrosarcoma and a fast-growing mass that accidentally mimicked teratoma. J Thorac Dis. 2016;8:E947-51.
- [CrossRef] [PubMed] [Google Scholar]
- Pulmonary extraskeletal myxoid chondrosarcoma: A case report and literature review. Int J Surg Case Rep. 2016;27:96-101.
- [CrossRef] [PubMed] [Google Scholar]
- A rare case of extraskeletal pulmonary myxoid chondrosarcoma. J Solid Tumors. 2015;6:18.
- [CrossRef] [Google Scholar]
- Giant primary mesenchymal chondrosarcoma of the lung: Case report and review of literature. Ann Thorac Cardiovasc Surg. 2013;19:481-4.
- [CrossRef] [PubMed] [Google Scholar]
- Primary dedifferentiated chondrosarcoma of lung: Report of a case. Zhonghua Bing Li Xue Za Zhi. 2011;40:127-8.
- [Google Scholar]
- Primary dedifferentiated chondrosarcoma of the lung. South Med J. 2009;102:861-3.
- [CrossRef] [PubMed] [Google Scholar]
- Primary chondrosarcoma of the lung with cutaneous and skeletal metastases. Singapore Med J. 2007;48:e196-9.
- [Google Scholar]
- Dedifferentiated chondrosarcoma of the lung: Case report and review of the literature. Clin Lung Cancer. 2007;8:439-42.
- [CrossRef] [Google Scholar]
- Primary chondrosarcoma of the lung recognized as a long-standing solitary nodule prior to resection. Jpn J Thorac Cardiovasc Surg. 2005;53:106-8.
- [CrossRef] [PubMed] [Google Scholar]
- Primary mesenchymal chondrosarcoma of the lung. Ann Thorac Surg. 2002;73:1960-2.
- [CrossRef] [Google Scholar]
- Primary pulmonary chondrosarcoma mimicking bronchogenic cyst on CT and MRI. Clin Imaging. 1996;20:181-3.
- [CrossRef] [Google Scholar]
- Primary chondrosarcoma of the lung. A clinicopathologic study. Cancer. 1993;72:69-74.
- [CrossRef] [Google Scholar]
- Primary mesenchymal chondrosarcoma of the lung. A case report with immunohistochemical and ultrastructural studies. Acta Pathol Jpn. 1992;42:364-71.
- [CrossRef] [Google Scholar]
- Fine-needle aspiration cytology of an unusual primary lung tumor, chondrosarcoma: Case report. Diagn Cytopathol. 1991;7:423-6.
- [CrossRef] [PubMed] [Google Scholar]
- Primary chondrosarcoma of the lung--a case report with immunohistochemical study. Jpn J Med. 1990;29:616-9.
- [CrossRef] [PubMed] [Google Scholar]
- Primary chondrosarcoma of the lung. A report of two cases. Clin Oncol. 1984;10:273-9.
- [Google Scholar]
- Primary chondrosarcoma of the lung. Case report and review of the literature. J Thorac Cardiovasc Surg. 1972;64:460-6.
- [CrossRef] [Google Scholar]
- Primary osteoid chondrosarcoma of the lung: Report of a case. Am J Cancer. 1933;18:603-9.
- [Google Scholar]




