
Translate this page into:
Hermansky-Pudlak Syndrome Complicated by Pulmonary Fibrosis: Radiologic-Pathologic Correlation and Review of Pulmonary Complications
-
Received: ,
Accepted: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Hermansky–Pudlak syndrome (HPS) is a rare autosomal recessive disorder characterized by oculocutaneous hypopigmentation, platelet dysfunction, and in many cases, life-threatening pulmonary fibrosis. We report the clinical course, imaging, and postmortem findings of a 38-year-old female with HPS-related progressive pulmonary fibrosis, highlighting the role of imaging in assessment of disease severity and prognosis.
Keywords
Hermansky–Pudlak syndrome
oculocutaneous hypopigmentation
platelet dysfunction
pulmonary fibrosis

INTRODUCTION

A 38-year-old female from Puerto Rico with complaints of hypoxia was transferred to our facility. She had been diagnosed 5 years earlier with Hermansky-Pudlak syndrome (HPS) subtype 1, complicated by pulmonary fibrosis. She had experienced a slow deterioration in pulmonary function, manifesting as worsening dyspnea, cough, and increasing oxygen dependence. In addition to pulmonary fibrosis, she had other manifestations of HPS, including multiple major bleeding episodes requiring blood transfusions, and oculocutaneous albinism with legal blindness. She required 24 h supplemental home oxygen with a baseline flow requirement of 6 l/min via nasal cannulae and was under evaluation for lung transplantation prior to her terminal hospital admission.
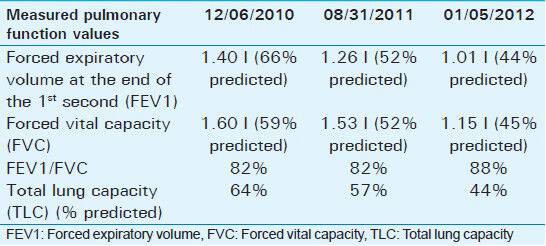
Pulmonary function tests [Table 1] showed increasing severe restrictive ventilatory deficit over the 2 years prior to admission, with total lung capacity going from 64% in 2010 to 57% in 2011 and 44% in 2012.
The patient presented with increasing dyspnea, pleuritic chest pain, and cough. Exercise capacity on admission measured via the 6-min walk test was 24% predicted. She was transferred to the intensive care unit due to hypoxia, with an oxygen saturation of 44% on 6 l nasal cannula. She was started on empiric antibiotics and supported with noninvasive ventilation, but continued to desaturate and was subsequently intubated.
The patient developed multiorgan failure as evidenced by elevated creatinine, transaminitis, and right heart failure. Blood and urine cultures on admission were negative, and white blood cell count was normal at 10.9 × 109 cells/L. Her oxygen saturation continued to drop with passive movement despite ventilator assistance. Given her poor prognosis, she was terminally extubated.
Radiologic features
Chest X-ray upon terminal admission demonstrated diffuse reticular interstitial opacities throughout the lungs [Figure 1]. Baseline CT of the chest obtained 16 months [Figure 2a] and 3 months before admission [Figure 2b] demonstrated progressive worsening of pulmonary fibrosis, as evidenced by increasing traction bronchiectasis, interlobular septal thickening, and consolidative opacities. Chest CT upon admission showed multifocal ground glass opacities, subpleural cysts, septal and peribronchovascular thickening, reticulation, and traction bronchiectasis involving greater than two-thirds of the lungs. Also, there was new honeycombing in the bilateral upper lobes suggesting end-stage fibrosis [Figure 2c].
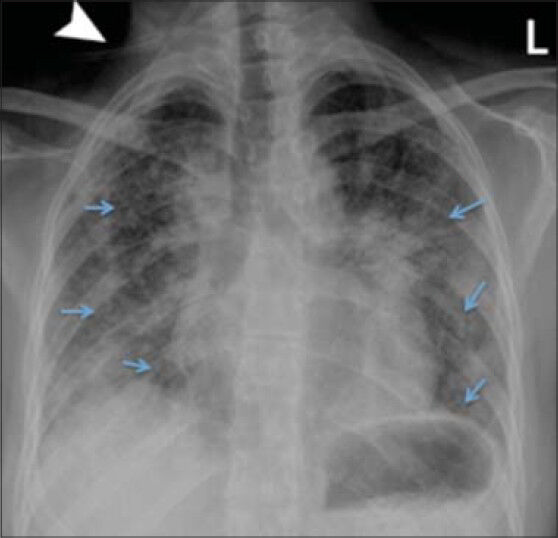
- 38-year-old female with a known history of Hermansky–Pudlak syndrome complicated by pulmonary fibrosis, admitted with hypoxia. Posteroanterior chest radiograph performed on admission demonstrates bilateral, mid to lower lung predominant, diffuse reticular interstitial opacities (arrows). Tubing from oxygen cannulation projects over the image (arrowhead).
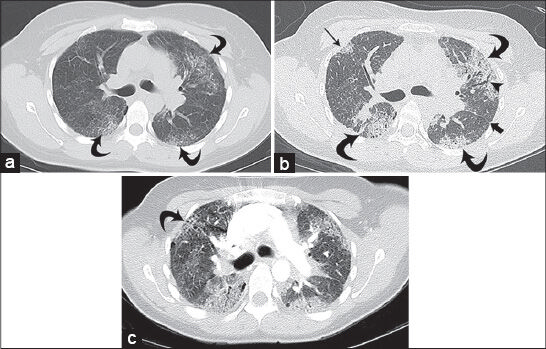
- 38-year-old female diagnosed with Hermansky–Pudlak syndrome complicated by pulmonary fibrosis, as evidenced by worsening dyspnea, cough, and increasing oxygen dependence. Baseline high-resolution CT (HRCT) of the chest: (a) Axial image on lung windows at the level of the carina demonstrates scattered, predominantly peripheral, ground glass opacities with associated fine peripheral reticulation in the lungs bilaterally (curved arrows); (b) high-resolution CT of the chest from the same level obtained 13 months later demonstrates progression of the peripheral opacities that have become more consolidative (curved arrows) with more conspicuous subpleural opacity in the right upper lobe (straight thin arrow); there is new bronchiectasis (arrowhead) and septal thickening along the left main fissure (thick arrow); and (c) axial CT of the chest on lung windows at terminal admission, performed as part of a CT pulmonary angiogram protocol (16 months after initial imaging), demonstrates further progression with increased size of peripheral consolidative opacities and increasing bronchiectasis. There is new honeycombing in the upper lobes bilaterally (curved arrow). Of note, the study did not demonstrate a pulmonary embolus.
Pathologic features
At postmortem examination, both lungs were found to be heavy with diffuse nodular parenchymal fibrosis [Figure 3a and 3b]. There was end-stage remodeling, characterized by cystic spaces lined by bronchial epithelium and surrounded by well-developed fibrosis on microscopic examination. The cysts, airspaces, and bronchioles were filled with mucus, suppurative exudate, and abundant vacuolated macrophages [Figure 4a and b].
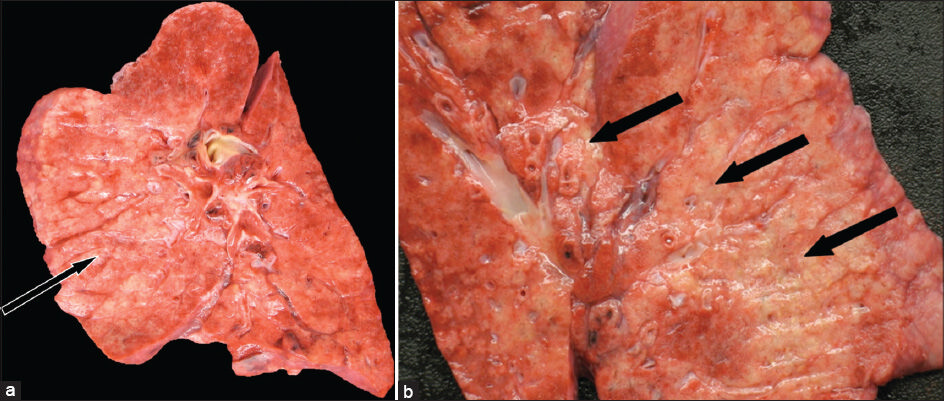
- 38-year-old female with history of Hermansky–Pudlak syndrome and end -stage pulmonary fibrosis, admitted with hypoxia and was terminally extubated after sustaining multiorgan failure. Postmortem autopsy was performed. (a and b) Photographs of coronal cut sections of a lung demonstrate diffuse nodular parenchymal fibrosis (arrows) with apparent bronchiolocentric distribution.
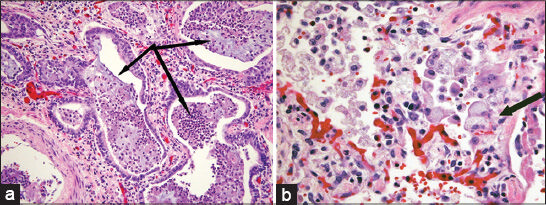
- 38-year-old female with history of Hermansky–Pudlak syndrome and end-stage pulmonary fibrosis, admitted with hypoxia. Photomicrographs of postmortem microscopic examination of lung tissue stained with hematoxylin–eosin stain: (a) ×200 magnification demonstrates end-stage remodeling, with honeycomb cystic spaces lined by metaplastic bronchial epithelium and surrounded by well-developed fibrosis (arrows); (b) ×400 magnification shows alveolar septal thickening associated with prominent diffuse vacuolization of type II pneumocytes (arrow).
DISCUSSION
HPS is a heterogeneous group of autosomal recessive disorders characterized by oculocutaneous hypopigmentation, platelet dysfunction (easy bruising and prolonged bleeding time despite normal platelet count), and lysosomal accumulation of ceroid-lipofuscin in the lungs, which primarily accounts for the associated morbidity.[1] In North America, most patients with HPS are from Puerto Rico, where the prevalence is estimated to be 1 in 1800 or occurring in 5 of every 6 albinos.[2] Groups of affected individuals have also been identified in Japan and a small village in Switzerland.[3] Patients usually present in childhood, often with recurrent epistaxis or prolonged bleeding after dental procedures.[4]
Nine subtypes of HPS are described, most of which are associated with a mutation in the HPS gene on the long arm of chromosome 10. Type 1 is the most common and most severe variant. This is also the most common subtype found in Puerto Rican patients.[2] This genotype leads to a high risk of pulmonary disease, hemorrhage, and, in approximately 15% of patients, granulomatous colitis.[4] Only Type 4 approaches Type 1 in severity, with the remaining subtypes behaving more mildly clinically and with little risk of restrictive lung disease.[2] While most patients with HPS1 die of pulmonary complications, approximately 13% die of complications from bowel inflammation, with this granulomatous colitis mimicking Crohn's disease and often occurring in adolescence or early adulthood.[4]
The pathophysiology is believed to be impaired intracellular trafficking of melanosomes, platelet dense bodies, and lysosomes.[5] Impaired formation of platelet-dense bodies leads to the bleeding dyscrasia, whereas impaired intracellular trafficking of melanin in the melanosomes of the skin and retina is postulated to be the cause of oculocutaneous albinism. Systemic complications are associated with accumulation of ceroid-lipofuscin, an amorphous lipid–protein complex in multiple organs and the reticuloendothelial system, leading to pulmonary fibrosis, granulomatous colitis, cardiomyopathy, and renal failure.[6] Ceroid-lipofuscin accumulation in the pulmonary alveolar macrophages is believed to be the primary mechanism for the development of pulmonary fibrosis. Recurrent hemorrhage with resulting hemosiderosis and inflammatory response is also suggested as an alternative mechanism.[7]
Pulmonary fibrosis in HPS is twice as common in women, occurring between the third and fifth decades.[8] It initially presents as dyspnea on exertion or restrictive lung disease on pulmonary function tests, and is the most common cause of death in affected patients.
The radiographic appearance of HPS is nonspecific and chest radiographs may be normal at presentation. Various radiographic abnormalities have been described, including reticular or reticulonodular opacities, pleural thickening, interstitial infiltrates, and perihilar fibrosis.[7] These findings may involve both lungs symmetrically or asymmetrically. Chest radiograph in our patient at the time of admission demonstrated symmetric reticular opacities throughout both lungs [Figure 1]. As chest radiographs sometimes underestimate the extent of disease, high-resolution CT is the imaging modality of choice for characterizing the parenchymal abnormalities and evaluating the extent of pulmonary involvement.
CT findings also vary depending on disease severity. Early stages of the disease are characterized by subtle reticulations, interlobular septal thickening, and peripheral ground glass opacities. In more advanced stages, there are more severe reticulations, peribronchovascular thickening, subpleural cysts, and bronchiectasis involving central airways.[67] Abnormalities on high-resolution CT tend to be evenly distributed throughout the lungs, with a slight predilection for the middle and lower lungs. Chest CT from our patient demonstrated diffuse septal thickening and ground glass opacities throughout both lungs, while honeycombing was predominantly seen in the bilateral upper lobes [Figure 2a–c]. HPS is more likely than the usual interstitial pneumonias or collagen diseases to also involve the upper lobes, particularly later in the course of the disease.[9]
Pulmonary hemorrhage, which typically presents with diffuse ground glass opacity on CT, should be considered in these patients given the underlying platelet dysfunction. Other entities that might have overlapping appearance on CT images include idiopathic pulmonary fibrosis (IPF), which features irregular reticulation, fibrosis and honeycombing in the posterior subpleural lung bases, and nonspecific interstitial pneumonitis (NSIP), which has bilateral and symmetric ground glass opacity and less honeycombing. However, IPF most commonly occurs in older patients and NSIP is not a progressive process. HPS has been classified by the American Thoracic Society and the European Respiratory Society as a mimic of IPF and one of the few diseases that has histological features similar, but not identical to usual interstitial pneumonia.[10]
A prior imaging study examining patients with HPS used a four-point score to grade the severity of CT appearance of disease. Grade 0 showed no changes. Grade 1 showed minimal changes (thickened interlobular septa, reticular disease, subpleural cysts, and areas of ground glass). Grade 2 had moderate disease (traction bronchiectasis, peribronchovascular thickening, and tracheal retraction in one-third or two-thirds of the lungs). Grade 3 had the findings of Grades 1 and 2, but involved more than two-thirds of the lungs. The study found that patients under 20 years of age typically had no CT findings (Grade 0) and those between 20 and 29 years had minimal CT changes (average grade 0.25 ± 0.16). Among patients older than 30 years, those with HPS1 mutations had significantly more severe disease on CT compared with those patients without this mutation (P = 0.001). Worsening CT appearance correlated highly with poorer pulmonary function; as CT severity score increased, the forced vital capacity (FVC) fell dramatically (P < 0.001). Also, higher CT severity score correlated with time to death, with over half the patients with Grade 3 disease dying within 4 months.[6] Our patient had a chest CT 16 months prior to admission, which showed Grade 2 findings [Figure 2a]. FVC at this time was 59% predicted. Subsequent chest CT 3 months before admission showed significant worsening of fibrosis, now involving greater than two-thirds of the lungs and meeting the criteria for Grade 3 disease [Figure 2b]. FVC at this time was 45% predicted. Final chest CT upon admission revealed extensive traction bronchiectasis, peribronchovascular thickening, and honeycombing, consistent with end-stage fibrosis [Figure 2c].
There is no effective treatment for pulmonary fibrosis due to HPS, besides lung transplantation. High-dose corticosteroids are administered to patients with advanced disease, though their efficacy has not been proven. Cigarette smoke and other pulmonary irritants must be avoided, and the antibiotic pirfenidone may slow the progression of pulmonary fibrosis in patients with significant residual lung. Supplemental oxygen is used in patients with dyspnea to alleviate discomfort.[6] Our patient had an objective and symptomatic decline over the last 12 months of her life, which rendered her oxygen dependent with a baseline requirement of 6 l/min and was under evaluation for lung transplantation prior to her terminal admission.
CONCLUSION
In summary, HPS is a rare congenital disorder, most often seen in Puerto Ricans, that manifests as easy bruising, epistaxis, prolonged bleeding, granulomatous colitis, and a high risk of pulmonary fibrosis. Patients with mutations in HPS1 are much more likely to develop more severe disease, typically after 30 years of age. CT correlates well with pulmonary function, and more severe CT changes significantly correlate with time to death. Transplantation remains the only current durable treatment for the disease.
ACKNOWLEDGMENTS
The authors would like to thank Dr. Michael Rosenthal, Dr. Nikhil Ramaiya, and Dr. Sree Harsha Tirumani of Dana Farber Cancer Institute and Dr. Scott Sheehan of Brigham and Women's Hospital for their contributions to this study.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2014/4/1/59/143437
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- Hermansky-Pudlak syndrome. Pulmonary manifestations of a ceroid storage disorder. Am J Med. 1979;66:737-47.
- [Google Scholar]
- Hermansky-Pudlak syndrome type 4 with interstitial pneumonia. J RMCR. 2013;9:38-41.
- [Google Scholar]
- Familial pulmonary fibrosis associated with oculocutaneous albinism and platelet function defect. A new syndrome. Q J Med. 1976;45:219-32.
- [Google Scholar]
- Pulmonary fibrosis in hermansky-pudlak syndrome. A case report and review. Respiration. 2006;73:382-95.
- [Google Scholar]
- Pulmonary function and high-resolution CT findings in patients with an inherited form of pulmonary fibrosis, Hermansky-Pudlak syndrome, due to mutations in HPS-1. Chest. 2000;117:129-36.
- [Google Scholar]
- Hermansky-Pudlak syndrome: Radiography and CT of the chest compared with pulmonary function tests and genetic studies. AJR Am J Roentgenol. 2002;179:887-92.
- [Google Scholar]
- The Hermansky-Pudlak syndrome: Radiographic features. Can Assoc Radiol J. 1986;37:42-5.
- [Google Scholar]
- Hermansky-pudlak syndrome: Report of a case and review of the literature. Int J Clin Exp Pathol. 2008;1:550-4.
- [Google Scholar]
- Hermansky-Pudlak syndrome with diffuse pulmonary fibrosis: Radiologic-pathologic correlation. J Comput Assist Tomogr. 1998;22:249-51.
- [Google Scholar]
- American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med. 2002;165:277-304.
- [Google Scholar]




