
Translate this page into:
Ganglioglioma of the Spinal Cord
Address for correspondence: Dr. Daniel C. Oppenheimer, Department of Imaging Sciences, University of Rochester Medical Center, 601 Elmwood Avenue, Rochester, NY 14642, USA. E-mail: Daniel_Oppenheimer@URMC.Rochester.edu
-
Received: ,
Accepted: ,
This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Ganglioglioma is a rare tumor consisting of neoplastic glial and neuronal elements. It accounts for only 0.5% of all primary central nervous system (CNS) neoplasms. We report an unusual case of extensive intramedullary thoracic spinal cord ganglioglioma in a 14-month-old girl who underwent subtotal resection followed by adjuvant chemotherapy. The epidemiology, histopathologic features, imaging findings, treatment, and prognosis are subsequently reviewed.
Keywords
Ganglioglioma
intramedullary
surgical debulking


INTRODUCTION
Ganglioglioma is a rare, slow-growing primary central nervous system (CNS) tumor which most frequently occurs in the temporal lobes, typically resulting in seizures.[1] Ganglioglioma of the spinal cord is exceedingly rare – only approximately 70 cases of spinal cord ganglioglioma have been reported, mostly in children or young adults.[2] The gold Standard of treatment is gross total resection; however, the optimal treatment in the setting of subtotal resection has not been clearly established.
CASE REPORT
A 14-month-old female child was initially evaluated for abnormal neck posture and difficulty turning her head to the right and upward. The patient was noted to perform compensatory maneuvers such as using her arms to push herself up to look around rather than simply lifting her head. Similarly, the patient was observed to make a 270° turn to the left rather than making a simple 90° turn to the right. Physical exam was notable for leftward deviation of the head with increased bulk and tone in the right sternocleidomastoid muscle with associated right shoulder elevation. Dextroconvex thoracic scoliosis was also present. Attempts at moving the neck provoked the patient to cry. She had good fine motor coordination for her age, but was not yet walking unassisted. No additional motor, sensory, gait, or reflex abnormalities were present. The physical exam finding of scoliosis prompted referral to orthopedic surgery and an MRI was subsequently obtained.
The MR revealed a long segment intramedullary lesion extending from T1 to T11 levels with corresponding cord expansion, heterogeneous enhancement following gadolinium administration [Figure 1], isointense T1 signal, and heterogeneously hyperintense T2 signal [Figure 2]. There was associated syringomyelia at the rostral and caudal aspects of the enhancing tumor. Additionally, there was T2 hyperintensity in the medulla and upper cervical spine as well as in the caudal spinal cord without associated enhancement.
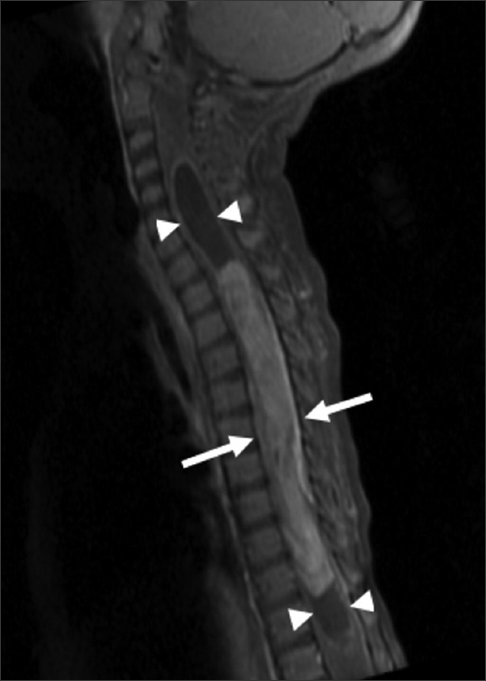
- 14-month-old female child with spinal cord ganglioglioma. Sagittal T1 fat-suppressed gadolinium contrast-enhanced MR image of the cervical and thoracic spine shows a long segment enhancing intramedullary lesion (arrows) extending from T1 to T11 with cord expansion and syringomyelia (arrowheads) at the rostral and caudal aspects of the enhancing tumor.
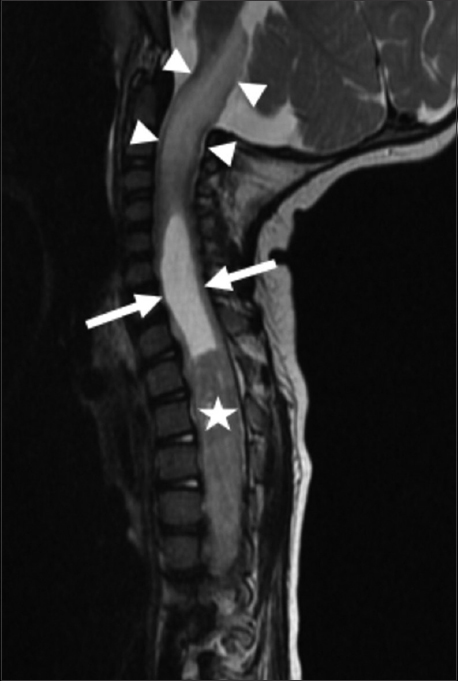
- 14-month-old female child with spinal cord ganglioglioma. Sagittal T2-weighted MR image of the cervical and thoracic spine shows syringomyelia (arrows) rostral to the intramedullary tumor (star). Abnormal hyperintense T2 signal and expansion is noted in the upper cervical spinal cord and brainstem (arrowheads).
The patient subsequently underwent thoracic en bloc T1–T12 laminectomy and debulking of thoracic intramedullary spinal cord tumor with use of electromyography, somatosensory evoked potentials, motor evoked potentials, and D-wave neuromonitoring. Tumor was debulked using a combination of posterior myelotomy and Cavitron Ultrasonic Surgical Aspirator (CUSA). Unfortunately, during exploration, it was determined that the tumor was variably interspersed with normal spinal cord. Therefore, only subtotal resection could be safely achieved with an intraoperative estimation of approximately 60–70% of the tumor resected.
Postoperatively, the patient maintained normal function of all extremities and MR showed reduced size of the enhancing tumor, less cord expansion, and improved T2 hyperintensity in the adjacent cord. However, due to the significant residual tumor burden, chemotherapy was initiated with Carboplatin and Vincristine. Unfortunately, follow-up imaging 3 months later showed increased enhancing intramedullary tumor with worsening cord expansion and T2 hyperintensity. Chemotherapy was changed to Vinblastine. Despite imaging evidence of progression of disease, the patient displayed no new neurologic deficits, although scoliosis and abnormal neck posture persisted. Additional follow-up MR 6 months after initiating Vinblastine showed improved cord edema and less-enhancing tumor. At the time of this writing, the patient is able to walk and jump unassisted, has started talking, and continues to meet appropriate developmental milestones.
Sections from the surgical excision revealed diffuse glial fibrillary acidic protein (GFAP)-immunoreactive glioma with collections of atypical, variably binucleate synaptophysin-immunoreactive cells with no Neu-N or neurofilament immunoreactivity [Figures 3–5]. There was focal necrosis and calcification, but no eosinophilic granular bodies. There was no immunoreactivity to isocitrate dehydrogenase gene 1 (R132) or p53. Cluster of differentiation 34 (CD34) immunostaining was confined to the blood vessels. The reticulin showed no notable pericellular staining. There were rare mitoses, but the Ki-67 labeling was variable and focally 15%. Polymerase chain reaction revealed no BRAF v600E mutation, NMYC or epidermal growth factor receptor gene amplification, and fluorescence in situ hybridization found no chromosome 10q loss. A diagnosis of glioneuronal tumor consistent with WHO grade I ganglioglioma was made. This diagnosis was confirmed with a second opinion obtained from a different academic institution.
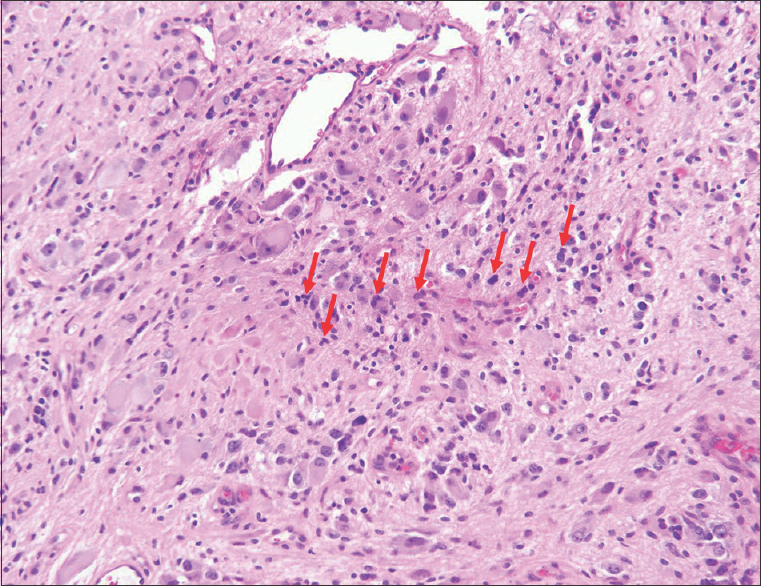
- 14-month-old female child with spinal cord ganglioglioma. Hematoxylin and eosin stained (×200).surgical resection specimen shows the tumor consisting of numerous dysplastic and binucleate ganglion cells in clusters (arrows).
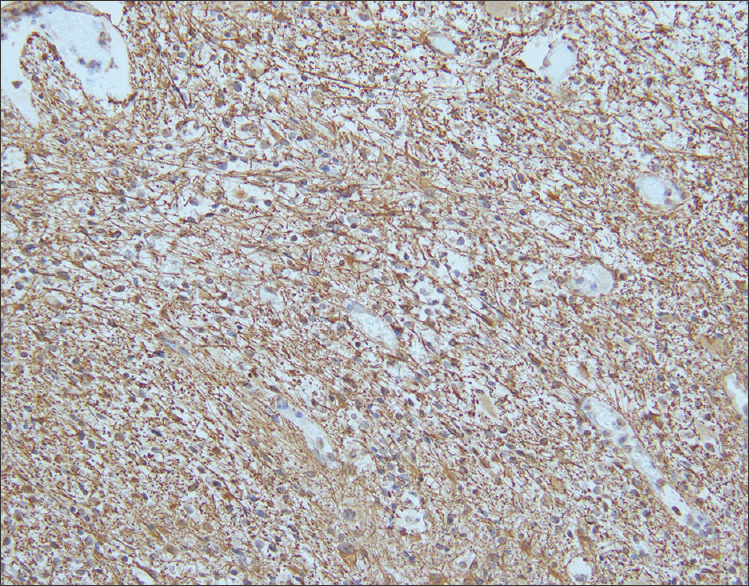
- 14-month-old female child with spinal cord ganglioglioma. Immunohistochemistry performed on the surgical resection specimen shows fibrillary GFAP-immunoreactive astrocytes (brown) spanning the tumor stroma (anti-GFAP immunohistochemical staining done with diaminobenzidine chromagen and hematoxylin counterstain; original magnification ×200).
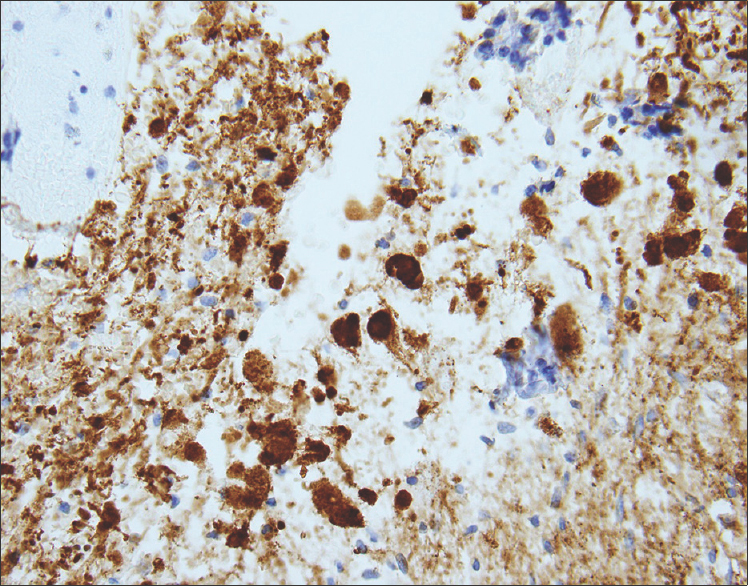
- 14-month-old female child with spinal cord ganglioglioma. Immunohistochemistry performed on the surgical resection specimen shows dysplastic and binucleate ganglion cells with extensive synaptophysin immunoreactivity in somas and processes (brown) (anti-synaptophysin immunohistochemical staining done with diaminobenzidine chromagen and hematoxylin counterstain; original magnification ×200).
DISCUSSION
Ganglioglioma is a rare CNS tumor consisting of neoplastic glial and neuronal elements.[1] The temporal lobe is the most frequent site of involvement; brainstem and spinal cord gangliogliomas are much less common. Within the spinal cord, ganglioglioma most frequently involves the cervical cord, although cases have been reported involving the thoracic and lumbar spine and the entire length of the spinal cord.[3] Spinal cord ganglioglioma has been observed in a wide age range of patients from newborn to elderly, but most cases occur in children or young adults. No gender, race, or ethnic association has been identified. Patients typically present with progressive myelopathy in addition to other neurological deficits related to location of the lesion. Motor and sensory defects, gait abnormalities, bowel and bladder dysfunction, and pain have all been described in patients with spinal ganglioglioma.[4]
Gangliogliomas are generally slow growing, and therefore, tumors are often large and detected at a late stage due to insidious progression. Only one case of spinal cord ganglioglioma presenting with acute neurological deficit has been described, which was attributed to tumor hemorrhage. In our case, the patient had several months of symptoms including scoliosis and neck deviation.
CT is generally not a recommended modality for diagnosis and evaluation of spinal cord tumors, particularly in the pediatric population. Evaluation with MR most commonly demonstrates a circumscribed solid or mixed solid and cystic mass spanning a long segment of the cord with hypointense T1 signal and hyperintense T2 signal in the solid component. Enhancement patterns are highly variable, ranging from minimal to marked, and may be solid, rim, or nodular. Adjacent cord edema and syringomyelia and peritumoral cysts may be present in addition to reactive scoliosis.[5] Our case demonstrated many typical imaging findings including long segment solid enhancing tumor, T2 hyperintensity, scoliosis, and syrinx formation.
It is nearly impossible to differentiate ganglioglioma from other more common intramedullary neoplasms based on imaging alone. Astrocytoma and ependymoma are more familiar intramedullary tumors which share many similar features to ganglioglioma, including T2 hyperintensity, enhancement, tumoral cysts, and cord edema. Poorly defined margins may be more suggestive of astrocytoma, while a central location in the spinal cord, hemorrhage, and hemosiderin staining are often seen with ependymoma. Hemangioblastoma and paraganglioma are less usual intramedullary tumors, but since they are more frequently encountered than ganglioglioma, they should also be included in the differential diagnosis.
Histologically, ganglioglioma is composed of both neoplastic glial and ganglion cells which are disorganized, variably cellular, and non-infiltrative. Occasionally, it may be challenging to differentiate ganglion cell tumors from an infiltrating glioma with entrapped neurons. The presence of neoplastic ganglion cells forming abnormal clusters, the presence of binucleation and dysmorphic neurons are helpful clues favoring ganglioglioma over glioma. The glial component of ganglioglioma includes fibrillary astrocytes with varying degrees of cellular atypia. The neoplastic neuronal components are often clustered or irregularly oriented. Fibrovascular stroma confined to the neuronal component, perivascular lymphocytic infiltrates, and small foci of calcification are common, as is immunopositivity for synaptophysin, neuron-specific enolase, and chromogranin A. Elevated Ki-67 and p53 labeling index is associated with more aggressive tumor behavior in both children and adults with gangliogliomas. The rare occurrence of malignant transformation is confined to the glial cell population, and is characterized by increased cellularity and mitotic activity, endothelial proliferation, and necrosis.[6]
Gangliogliomas are generally benign WHO grade I tumors; the presence of anaplastic changes in the glial component is considered to represent WHO grade III (anaplastic ganglioglioma). Criteria for WHO grade II have been suggested, but are not established.[7] Malignant transformation of spinal ganglioglioma has been seen in only a select few cases.[3] Poor prognostic factors for adults with gangliogliomas include older age at diagnosis, male sex, and malignant histologic features.
Definitive treatment for ganglioglioma requires gross total surgical resection, and a good prognosis is generally expected when this is achieved.[8] However, indistinct tumor margins and the desire to preserve normal spinal cord tissue, motor and sensory function may preclude complete resection of tumor. According to a series by Lang et al., reviewing several patients with resected spinal cord ganglioglioma, the 5- and 10-year survival rates after total resection were 89% and 83%, respectively.[4] In that study, patients with spinal cord ganglioglioma had a 3.5-fold higher relative risk of tumor recurrence compared to patients with supratentorial ganglioglioma. It has been recognized that postoperative results correlate closely with preoperative neurological status as well as the ability to achieve complete resection.[9] With the exception of WHO grade III anaplastic ganglioglioma, radiation therapy is generally regarded to have no role in the treatment of ganglioglioma.[10] In fact, radiation therapy may induce malignant transformation of a recurrent ganglioglioma several years later. Adjuvant chemotherapy is also typically reserved for anaplastic ganglioglioma, but has been used anecdotally in partially resected low grade spinal cord gangliogliomas which show evidence of disease progression, such as in the case presented above.
CONCLUSION
We present a case of extensive thoracic spinal ganglioglioma in a 14-month-old girl, whose clinical symptoms remained stable after subtotal resection and adjuvant chemotherapy, despite imaging evidence of tumor growth. While ganglioglioma is a very rare tumor of the spinal cord, it must be considered in the differential diagnosis of an intramedullary tumor. Early recognition, diagnosis, and treatment is imperative to improve prognosis and minimize neurological sequelae. Unfortunately, the diagnosis is often delayed in clinical practice due to the slow growth and indolent nature of this tumor. The benefit of radical resection must be weighed against the risk of further worsening neurological function, particularly in young patients who have a longer life expectancy. Further research on the optimal treatment of partially resected and recurrent spinal cord ganglioglioma is necessary.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2015/5/1/53/166355
REFERENCES
- Childhood ganglioglioma and medically intractable epilepsy.A clinicopathological study of 15 patients and a review of the literature. Pediatr Neurosurg. 1995;22:181-8.
- [Google Scholar]
- Ganglioglioma of the spinal cord: Report of two rare cases and review of the literature. Neurosurgery. 1997;41:1410-6.
- [Google Scholar]
- Central nervous system gangliogliomas. Part 2: Clinical outcome. J Neurosurg. 1993;79:867-73.
- [Google Scholar]
- Ganglioglioima: An ultrastructural and immunohistochemical study. Cancer. 1997;79:989-1003.
- [Google Scholar]
- WHO Classification of Tumours of the Central Nervous System. (4th ed). Lyon: International Agency for Research on Cancer; 2007. p. :103.
- [Google Scholar]
- Pediatric low-grade ganglioglioma: Epidemiology, treatments, and outcome analysis on 348 children from the surveillance, epidemiology, and end results database. Neurosurgery. 2015;76:313-20.
- [Google Scholar]
- Clinical outcome of pediatric gangliogliomas: Ninety-nine cases over 20 years. Pediatr Neurosurg. 1997;27:203-7.
- [Google Scholar]
- Ganglioglioma of the spinal cord: Report of two cases and review of literature. J Clin Neurosci. 2004;11:199-203.
- [Google Scholar]




