
Translate this page into:
Crouzon Syndrome: Clinico-Radiological Illustration of a Case
Address for correspondence: Dr. Raviprakash Sasankoti Mohan, C/o Dr. R. P. Singh, Dhanwantri Nursing Home, Sarai Khalsa, Behind Head Post Office, Moradabad - 244 001, Uttar Pradesh, India. E-mail: sasan_ravi@rediffmail.com
-
Received: ,
Accepted: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Crouzon syndrome, also called craniofacial dysostosis, is an autosomal dominant disorder with complete penetrance and variable expressivity. Described by a French neurosurgeon in 1912, it is a rare genetic disorder characterized by premature closure of cranial sutures, midfacial hypoplasia, and orbital defects. Here, we report a case of this rare entity. The patient presented with brachycephaly, maxillary hypoplasia, exophthalmos, mandibular prognathism, along with dental and orbital abnormalities.
Keywords
Craniofacial dysostosis
crouzon syndrome
copper beaten appearance
exophthalmos

INTRODUCTION

Crouzon syndrome (CS) is a rare genetic disorder characterized by premature closure of one or more cranial sutures and produces the characteristic craniofacial and other associated abnormalities.[12] It is one of the craniosynostosis syndrome that is caused by a mutation in the fibroblast growth factor receptor 2 gene (FGFR2).[2] It accounts for approximately 4.8% of all cases of craniosynostosis making it the most common syndrome within the craniosynostosis group. It has worldwide prevalence rate of approximately 1 per 25,000 live births.[1–3] Once the sutures close, growth potential of those sutures is restricted. However, multiple sutural synostoses frequently extend to premature fusion of skull base causing midface hypoplasia, shallow orbit, maxillary hypoplasia, and occasional upper airway obstruction.[4] Other clinical features include hypertelorism, exophthalmos, strabismus, beaked nose, short upper lip, maxillary hypoplasia, and relative mandibular prognathism with no digital abnormalities.[5–7] CS is distinguishable from other craniosynostosis syndromes by lack of hand and/or foot abnormalities.[1] In this article, we present a case of CS in a 20-year-old female patient.
CASE REPORT
A 20-year-old female along with her parents came to the department of oral medicine with a complaint of forward placement of the lower jaw with respect to the upper jaw. As the patient's appearance and head size was not normal, a detailed family and medical history was taken. Review of medical history was unremarkable. The mother reported normal labor and delivery. There were no anomalies in any siblings or near relatives. The enlarged size of the head was noticed by the mother when the girl was 9 months old and the severity had gradually increased.
On general examination, the patient was of short stature, had a wide nasal bridge, hypertelorism, and exophthalmos. Intra-oral examination revealed high arch palate, hypoplastic maxilla causing prognathism of the mandible, and class III malocclusion [Figure 1a–d]. The patient was evaluated by an opthalmologist for complaints regarding vision. The findings revealed papilloedema suggestive of raised intracranial pressure.
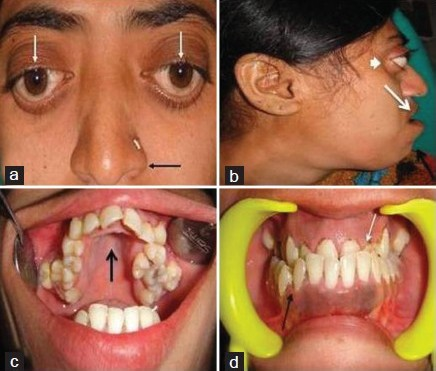
- Patient photograph shows (a) hypertelorism (white arrow), parrot beak nose (black arrow). (b) Profile view shows hypoplastic maxilla (white arrow) leading prognathism of mandible (black arrow) and exophthalmos (smallwhitearrow). (c) Intra oral view shows high arch palate (black arrow). (d) Front view of the teeth reveal hypoplastic maxilla (white arrow) and class III malocclusion (black arrow).
The patient was subjected to radiographic investigations. The orthopantomogram revealed the presence of all permanent teeth with no obvious abnormality. Lateral skull view demonstrated a retruded maxilla with a relatively large mandible and copper beaten appearance [Figure 2]. On anteroposterior spine radiograph, decreased intervertebral space between C5 and C6 vertebrae was seen [Figure 3]. Radiographic examination of metacarpal bones and fingers were unremarkable. Paranasal sinus view revealed obliterated sutures, small paranasal sinuses, and prominent cranial markings of the inner surface of cranial vault seen as multiple radiolucencies appearing as depressions (digital impressions) [Figure 4]. Three-dimensional (3D) computed tomographic images of the skull revealed increased circumference of the skull and moderate degree of hydrocephalus with diffuse indentation of inner table of the skull [Figures 5 and 6].
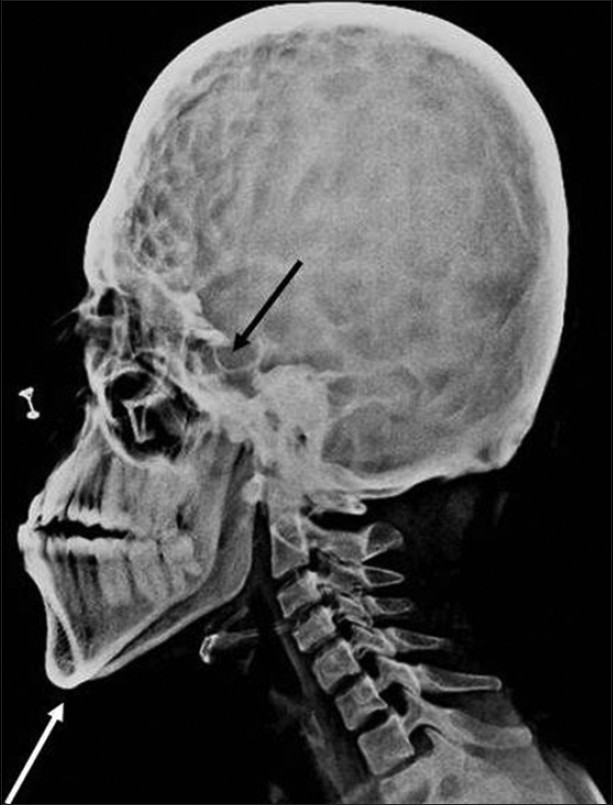
- Lateral skull projection reveals mandibular prognathism (white arrow), maxillary hypoplasia, copper beaten appearance, and enlarged hypophyseal cavity (black arrow).
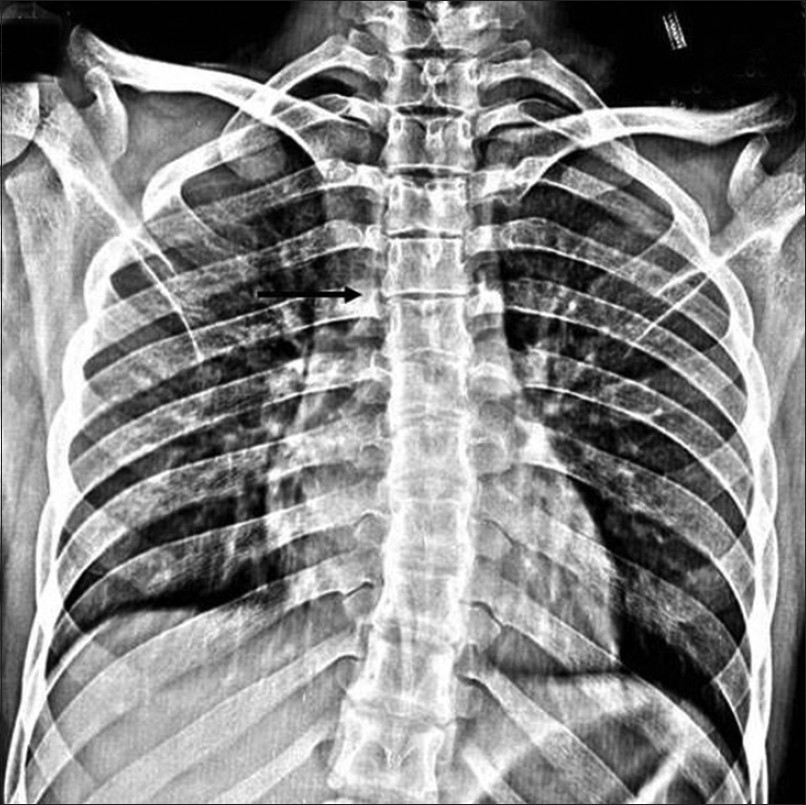
- Anteroposterior spine radiograph shows decreased intervertebral space between C5 and C6 (black arrow).
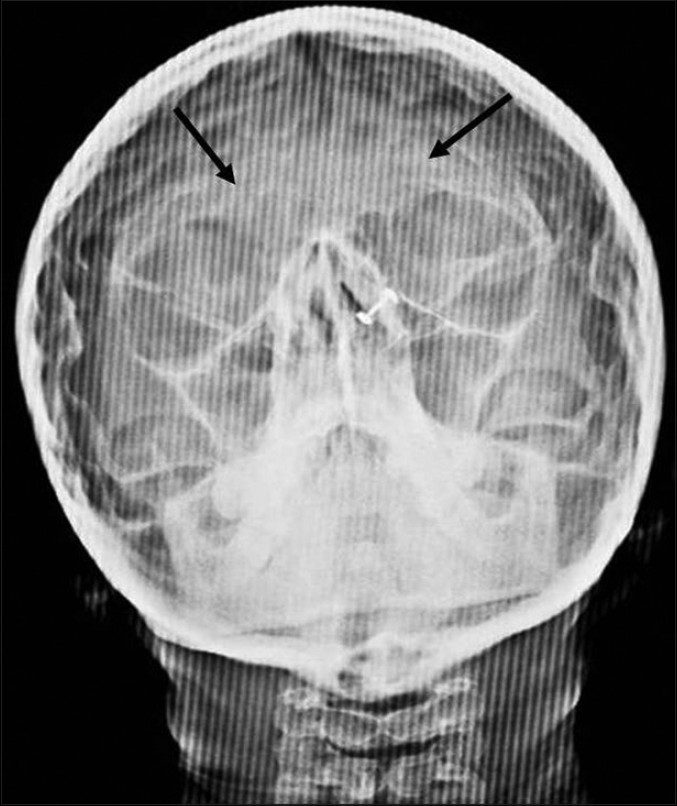
- Paranasal sinus view shows prominent convolution markings suggestive of copper beaten appearance (black arrow).
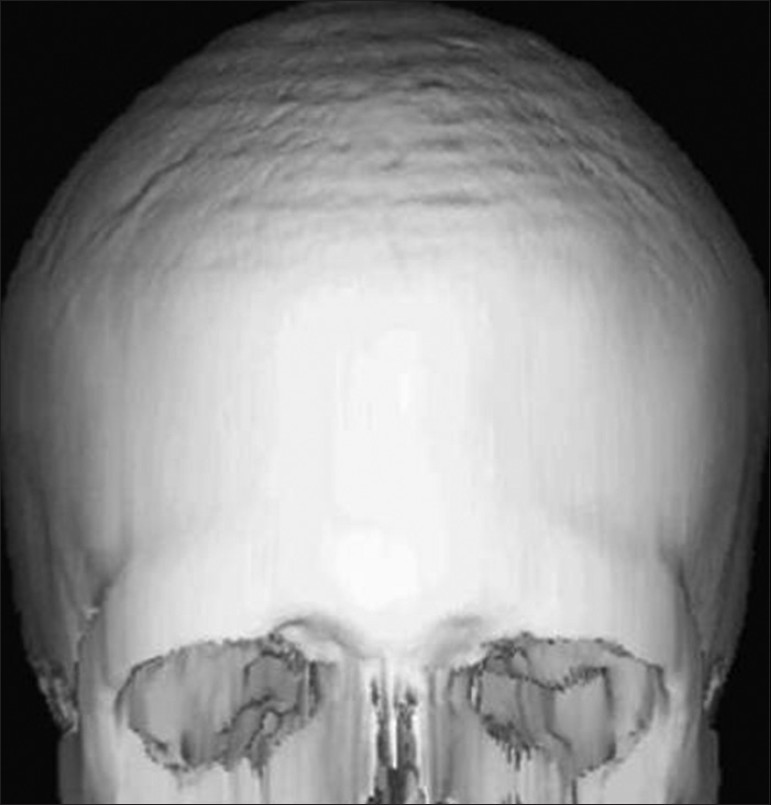
- Three-dimensional (3D) computed tomographic images of skull show increased circumference of the skull
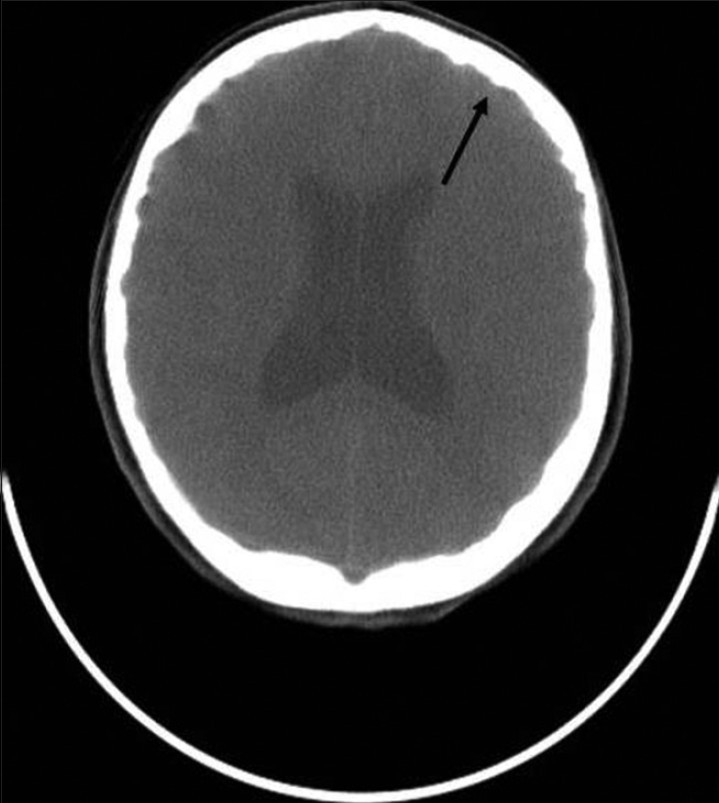
- Computed tomography (CT) image of the skull shows moderate degree of hydrocephalus with diffuse indentation of inner table of skull (black arrow).
The clinical, dental, ophthalmologic features, and radiographic findings pointed to the diagnosis of Crouzon syndrome.
Owing to the compromised oral hygiene, a thorough professional oral prophylaxis was performed. The patient was then referred to the department of orthodontics and dentofacial orthopedics for the correction of maxillary hypoplasia, mandibular prognathism, and class III malocclusion.
DISCUSSION
In 1912, a French neurologist, Octave Crouzon (1874 - 1938) first described the hereditary syndrome of craniofacial synostosis, which includes a triad of skull deformities, facial anomalies, and exophthalmos and is now known as Crouzon syndrome.[12] Similar characteristic features of varying degrees were evident in our patient. CS is an autosomal dominant disorder with complete penetrance and variable expressivity.[18] This syndrome is caused by mutations in the fibroblast growth factor receptor-2 (FGFR2) gene which is mapped to chromosome locus 10q25 - 10q26, but exhibits locus heterogeneity with causal mutations in FGFR2 (CS) and FGFR3 (CS with acanthosis nigricans) in different affected individuals.[1246]
CS has no racial or sex predilection.[4] However, when the craniosynostosis is of sagittal or metopic types, the predominance increases in boys, while coronal craniosynostosis is more common in girls.[12] The condition is usually detected in the first year of life.[2] However, there are also congenital premature forms in which the synostosis begins inside the uterus and is evident at birth with facial deformities.[3] Premature closure of cranial sutures most commonly the coronal and sagittal ones results in abnormal skull growth and affects the growth and development of the orbits and maxillary complex. Other clinical features include hypertelorism, exophthalmos, strabismus, beaked nose, short upper lip, maxillary hypoplasia, and relative mandibular prognathism with no digital abnormalities.[5–7]
Thorough clinical and radiological analyses are required for early recognition and diagnosis of CS. The radiographs reveal obliterated sutures, hypoplastic maxilla with shallow orbits, shortened cranial fossa, enlarged hypophyseal cavity, and small paranasal sinuses.[238] Prominent cranial markings of the inner surface of cranial vault may be seen as multiple radiolucencies appearing as depressions resulting in hammered silver/beaten metal/copper beaten appearance.[4] A copperbeaten skull was seen in the radiograph of our patient indicating internal remodeling of the calvaria due to an increase in intracranial pressure, as a result of premature cranial suture fusions.[3] On spine radiograph, abnormal craniocervical junction, butterfly-shaped vertebrae, and fusion of cervicalvertebrae (C2-C3 and C5-C6) may be visible.[8] Radiographic examination of metacarpal bones and fingers are unremarkable.[45]
CS must be differentiated from simple craniosynostosis and other syndromic craniosynostosis. Differential diagnosis is made with other syndromes associated with features of craniosynostosis such as Pfeiffer's syndrome, Apert syndrome, Saethre–Chotzen syndrome, Carpenter syndrome, and Jackson–Weiss syndrome.[24] All these involve craniofacial abnormalities, as well as other abnormalities including the hands or feet. Craniosynostosis may also be caused by abnormal external forces such as decreased brain growth or abnormal fetal head positioning, and in these patients, abnormal head shape may correct itself with time.[9]
The management of CS requires a multidisciplinary approach for successful outcome. The goal is to stage reconstruction to coincide with facial growth patterns, visceral function, and psychosocial development.[8] Surgical treatment varies according to the variable expressivity of the disease and usually begins during a child's first year with fronto-orbital advancement with cranial decompression.[10] Subsequent development of midfacial hypoplasia needs correction. Procedures for this purpose include the Le Fort III osteotomy or its segmental variants, monobloc frontofacial advancement, or bipartition osteotomy. Early craniectomy with frontal bone advancement is most often indicated to prevent or treat increased intracranial pressure because newborns with Crouzon syndrome develop multiple suture synostoses and fused synchondroses. Fronto-orbital and midfacial advancements help in the cosmetic reconstruction of facial dysmorphisms. A new technique, craniofacial disjunction, followed by gradual bone distraction (Ilizarov procedure) has been reported to produce complete correction of exophthalmos and improvement in the functional and esthetic aspects of the middle third of the face without the need for bone graft in patients aged 6-11 years. Adult CS, as in our case presenting with marked midface hypoplasia and exorbitism, can be corrected by orbital decompression and zygomaticomaxillary advancement.[910]
The prognosis depends on the severity of malformations and the timing of intervention. Innovations in craniofacial surgery have enabled patients to achieve their full potential by maximizing their opportunities for intellectual growth, physical competence, and social interaction.[79]
CONCLUSION
Crouzon syndrome should be managed as early as possible as it results in impaired facial appearance and other complications like mental retardation, airway obstruction, and decreased visual acuity as the patient gets older. With proper treatment, these patients can be productive and active members of the main stream of society.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2012/2/1/70/104303
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- Dentofacial features of a family with Crouzon syndrome. Case reports. Aust Dent J. 1997;42:11-7.
- [Google Scholar]
- Crouzon's syndrome: A review of literature and case report. Contemp Clin Dent. 2011;2:211-4.
- [Google Scholar]
- Opthalmological and radiological picture of crouzon syndrome: A case report. Acta Medica Medianae. 2009;48:37-40.
- [Google Scholar]
- Crouzon syndrome. A case report and review of literature. Indian J Otolaryngol Head Neck Surg. 2006;58:381-2.
- [Google Scholar]
- Crouzon's syndrome literature review. Intt Arch Otorhinolaryngol Sao Paulo. 2008;12:436-41.
- [Google Scholar]
- The value of the maxillo-malar osteotomy in the treatment of Crouzon syndrome with exorbitism. Ann Plast Surg. 2008;61:285-9.
- [Google Scholar]




