
Translate this page into:
Clinico-radiologic Findings in Group II Caudal Regression Syndrome
Address for correspondence: Dr. Pankaj Sharma, (Presently affiliated to Delhi State Cancer Institutes, Delhi) B 78, Sector 23, Noida - 201 301, Uttar Pradesh, India. E-mail: pankajrad7477@yahoo.com
-
Received: ,
Accepted: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Caudal regression syndrome (CRS) is a rare congenital abnormality in which a segment of the lumbo-sacral spine and spinal cord fails to develop. The severity of the morphologic derangement inversely correlates with residual spinal cord function. We present a case report of a 10-year-old girl with Group 2 CRS, to emphasize clinical and radiologic findings in this rare abnormality.
Keywords
Dysgenesis
dysraphic
imperforate
lipomyelocystocele


INTRODUCTION
Caudal regression syndrome (CRS) is a spectrum of disorders of caudal vertebral agenesis or dysgenesis, often with spinal cord malformations that is associated with other congenital anomalies, especially of the genitourinary and gastrointestinal systems.[1]
The incidence of CRS is 1 to 5 cases in 100,000 births. The cause of CRS is largely unknown, but in 16% of the cases, the syndrome is associated with maternal diabetes.[2] We present a rare case of Group 2 CRS in a 10-year-old girl with associated findings including anterior epidural lipoma, anterior lipomyelocystocele, an irregular trabeculated urinary bladder, bilateral hydronephrosis, and bilateral hydroureter.
CASE REPORT
A 10-year-old girl presented with progressive lower extremity weakness, difficulty in walking, and urinary incontinence. She had urinary incontinence since infancy and had undergone surgery for neurogenic bladder 5 years earlier. She now had progressively increasing urinary incontinence,, increased weakness of the lower limbs, and bowel incontinence. Her legs showed wasting of both proximal and distal muscles. Scoliotic deformity was seen along with impairment in walking. No cutaneous stigmata were noted in the back. Blood investigation findings were normal. She underwent technetium dimercaptosuccinic acid scans, which revealed bilateral atrophic kidneys with impaired renal function and parenchymal scarring.
Magnetic resonance imaging (MRI) examination of the lumbosacral spine revealed scoliotic deformity, bilateral sacral agenesis (S2 level), low lying spinal cord (L5 level), and syringohydromelia of the caudal spinal cord. Associated findings include anterior epidural lipoma, anterior lipomyelocystocele, an irregular trabeculated urinary bladder, bilateral hydronephrosis, and bilateral hydroureter [Figures 1-3]. Based on the clinical and radiologic findings, a diagnosis of CRS Group 2 was made.
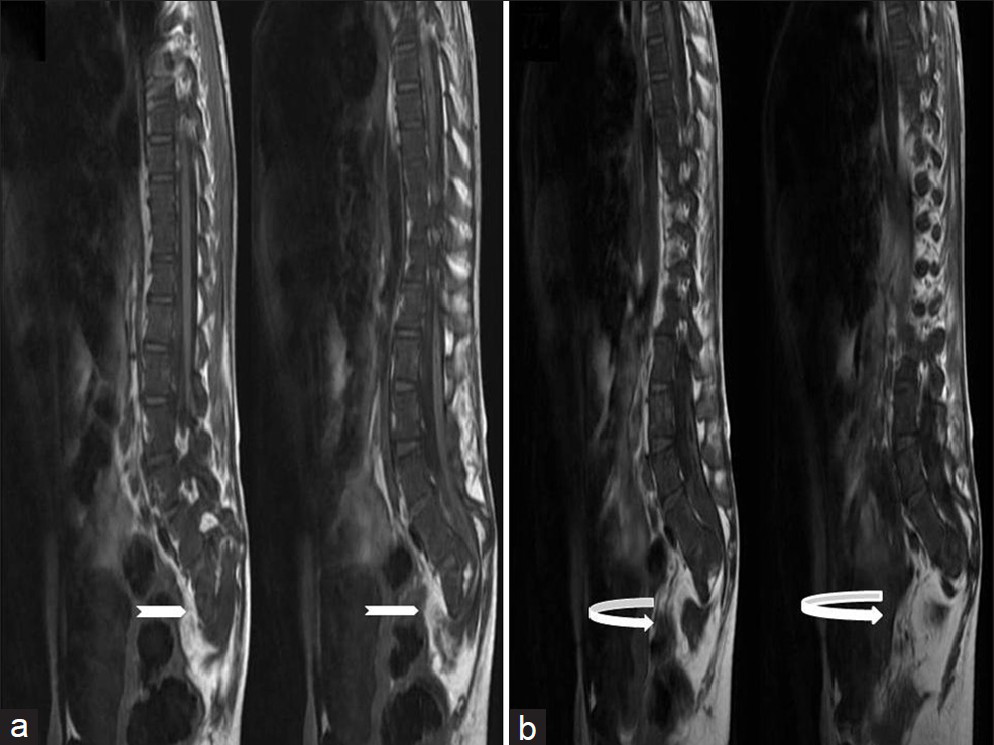
- Ten-year-old girl diagnosed with Group II Caudal regression syndrome. (a and b) Sagittal T1 weighted images show bilateral sacral agenesis (white arrow, S2 level) with anterior lipomyelocystocele (curved white arrow).
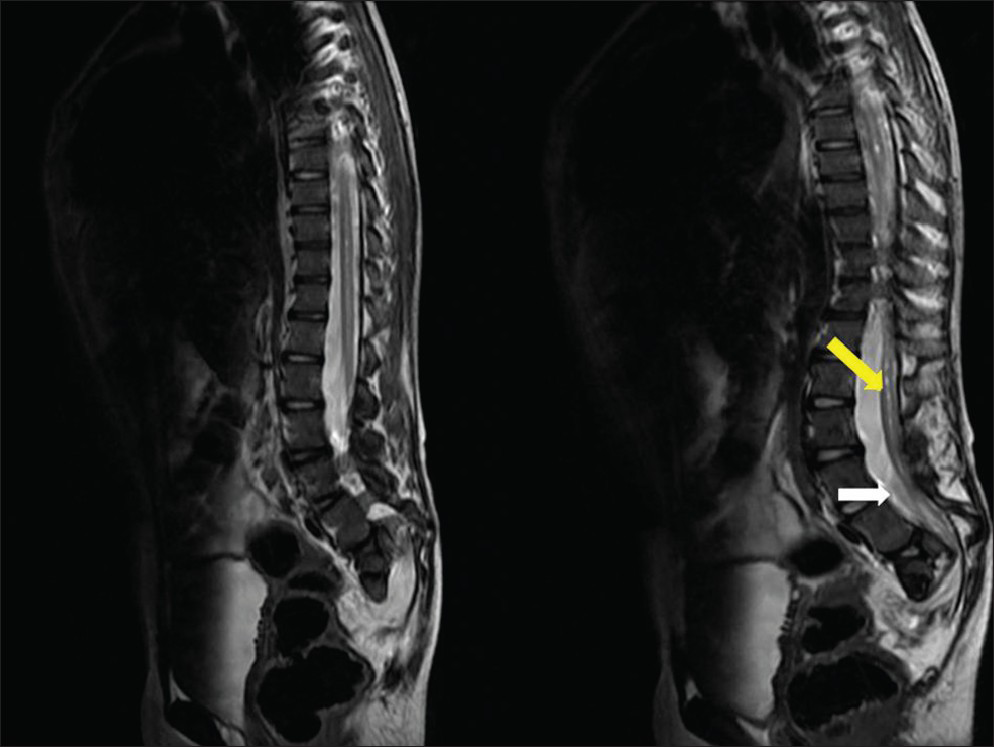
- Ten-year-old girl diagnosed with Group II Caudal regression syndrome. Sagittal T2 weighted image shows low lying spinal cord (white arrow, L5 level) with syringohydromelia (pointed yellow arrow) of the caudal spinal cord.
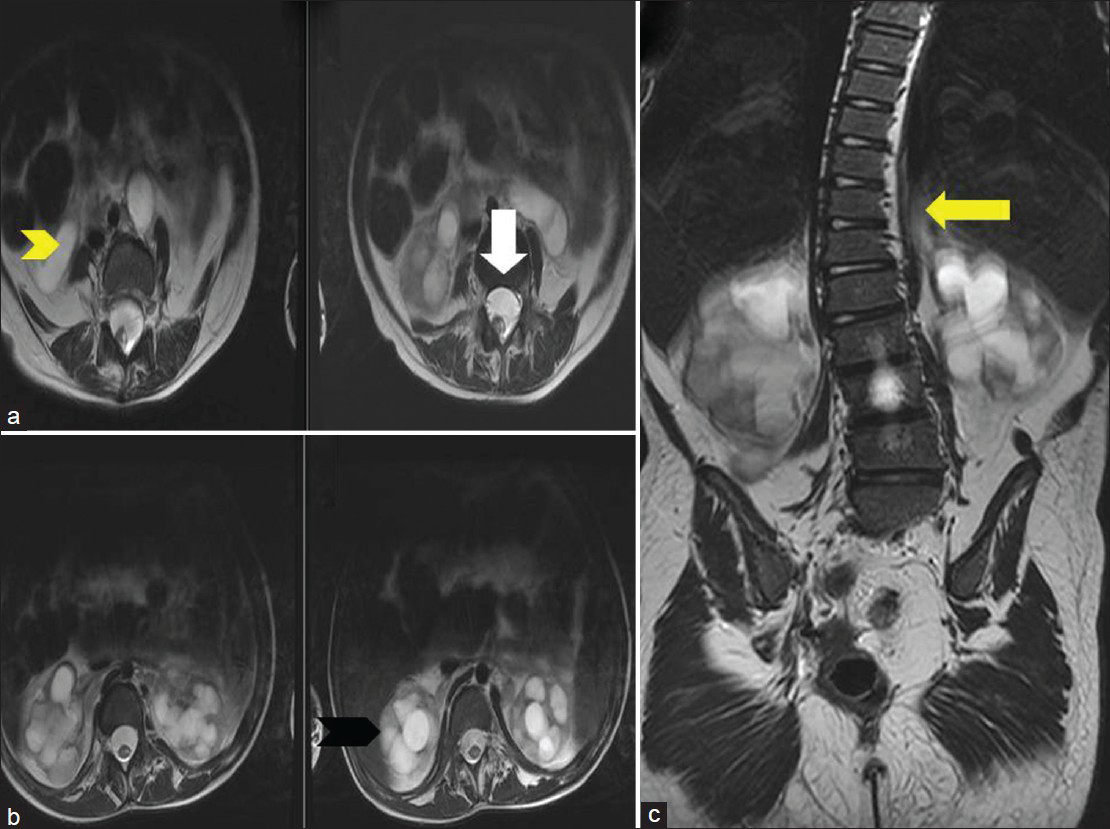
- Ten-year-old girl diagnosed with Group II Caudal regression syndrome. (a) Axial T2 weighted image shows anterior epidural lipoma (white arrow). (b) Axial T2 weighted image shows bilateral hydronephrosis (black arrowhead) and bilateral hydroureter (yellow arrowhead). (c) Coronal T2 weighted image reveals lumbosacral scoliosis (yellow arrow).
DISCUSSION
CRS is a rare disorder characterized by abnormal development of the caudal spine of the developing fetus. Abnormalities in CRS range from absent coccyx to sacral or lumbar agenesis. In extreme cases, last intact vertebrae are T11 or T12. The most characteristic feature of the spinal cord terminus in CRS is the wedge shaped cord terminus, that is the dorsal aspect of the cord extending more caudally than the ventral portion.[34] When this characteristic cord terminus is seen, imaging of the lower lumbar and sacral regions should be performed to verify the diagnosis of caudal regression.
Neurologic anomalies in CRS include segmental deficit corresponding to the level of vertebral agenesis, neurogenic bladder, bowel incontinence or obstipation, dimples and absent or deep tendon or skin reflexes.[3] Neurologically, lumbosacral sensation are much better preserved than the motor functions and urinary and bladder symptoms are unusual.[5] The level of vertebral agenesis correlates with motor level but not the sensory level.
Orthopedic anomalies in CRS include caudal vertebral agenesis, dysraphic or dysplastic vertebral defects, hip dislocation or dysplasia, clubfeet, frog leg position, narrow pelvis, and sirenomelia.[3] Genitourinary anomalies in CRS include renal dysplasia or agenesis, hydronephrosis, dysplasia or agenesis of other parts of the genitourinary system and renal ectopia.[3] Gastrointestinal anomalies in CRS include imperforate anus or anorectal atresia, fistulas and esophageal or duodenal atresia.[3]
According to Smith et al.,[6] and Karrer et al.,[7] caudal vertebral and spinal cord anomalies occur in 40% to 50% of children with anorectal malformations. Hence, special attention should be paid to caudal regression in these children. MRI screening should be ordered for patients with characteristic features or X-ray evidence of sacral agenesis, anorectal and genitourinary anomalies, or other complex malformations of the caudal region.[5] MRI enables adequate diagnosis not only of the congenital anomalies of the spinal cord and spine, but also of most associated anomalies in the pelvic region.
CRS was first proposed as a clinical entity by Duhamel in 1960 in a meeting of the British Association of Pediatric Surgeons. Sirenomelia is a rare and lethal congenital anomaly, caused by a disruptive vascular defect, characterized by fusion of the lower extremities, associated with bilateral renal agenesis, absence of the sacrum, rectum, and bladder. Sirenomelia was formerly thought to be an extreme form of CRS, but it is reclassified to be considered a separate entity. Aside from fusion of the lower extremities, presence of single umblical, persistent vitelline artery is the chief distinguishing anatomic finding from CRS.
The etiology of Sirenomelia and CRS is unknown.[8910] Diversion of blood flow away from the caudal portion of the embryo through the abdominal umbilical artery/“vascular steal” has been proposed as the primary mechanism leading to Sirenomelia. In contrast, CRS is hypothesized to arise from primary defect of caudal mesoderm. A teratogenic event during the gastrulation stage i.e. 3rd gestational week, may interfere with the formation of notochord, resulting in abnormal development of caudal structures. Altered oxidative metabolism from maternal diabetes may cause increased production of free oxygen radicals in the developing embryo, which may be teratogenic.
Based on position of conus, Pang,[5] divided patients into two groups:
-
Group 1: Conus ends cephalic to the lower border of L1 vertebrae. In these patients, sacral deficit is large ending at or above S1 vertebrae.
-
Group 2: Conus ends caudal to the lower border of L1 vertebrae. In these patients, sacrum tends to be well-preserved with identifiable portions of S1 or lower vertebrae. Conus is elongated, stretched caudally and tethered by thick filum terminale (65%), terminal myelocystocoele (15%), terminal hydromyelia (10%) and transitional lipoma.
Pang concluded dural stenosis and tethering lesions to be much more common in patients with sacral agenesis than previously thought. Paradoxically, tethered cord was found more commonly in mild versus severe sacral agenesis. Neurologically, deterioration was frequently associated with tethered cord and in dural stenosis, but not with blunt, short conus. Hence, all tethered lesions should be excised to release the cord and dural stenosis should be treated by decompressive duroplasty.
In our case, child had Group 2 CRS with conus ending at L5 vertebral level and sacrum ending at S2 level. Associated findings included syringohydromelia of the caudal spinal cord, anterior epidural lipoma, anterior lipomyelocystocele, an irregular trabeculated urinary bladder, bilateral hydronephrosis, and bilateral hydroureter.
The prognosis for children with CRS depends on the severity of the lesion and the presence of associated anomalies. Surviving infants have usually normal mental function and they require extensive urologic and orthopedic assistance. Surgical untethering, release or duraplasty are procedures that may be necessary to improve neurological function.
CONCLUSION
CRS is a rare congenital abnormality, which has distinctive clinical and radiological features. MRI not only helps in confirming the diagnosis, but also has an important role in identifying associated abnormalities and further management.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2013/3/1/26/114214
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- Caudal aplasia syndrome. In: Vinken PJ, Bruyn GW, eds. Handbook of Clinical Neurology. Part 32. 1978. p. :347-58.
- [Google Scholar]
- Classification of congenital abnormalities of the CNS. AJNR Am J Neuroradiol. 1988;9:315-26.
- [Google Scholar]
- MR of the caudal regression syndrome: Embryologic implications. AJNR Am J Neuroradiol. 1994;15:1021-9.
- [Google Scholar]
- The wedge-shaped cord terminus: A radiographic sign of caudal regression. AJNR Am J Neuroradiol. 1989;10:1223-31.
- [Google Scholar]
- Sacral agenesis and caudal spinal cord malformations. Neurosurgery. 1993;32:755-78.
- [Google Scholar]
- Associated anomalies. In: Stephens FD, Smith ED, eds. Anorectal Malformations in Children: Update 1988, Birth Defects. Vol 24. New York: Liss C; 1988. p. :501-49.
- [Google Scholar]
- Anorectal malformations: Evaluation of associated spinal dysraphic syndromes. J Pediatr Surg. 1988;23:45-8.
- [Google Scholar]
- Sirenomelia, the mermaid syndrome – Detection in the first trimester. Prenat Diagn. 2003;23:493-5.
- [Google Scholar]
- Five cases of caudal regression with an aberrant abdominal umbilical artery: Further support for a caudal regression-sirenomelia spectrum. Am J Med Genet A. 2007;143A:3175-84.
- [Google Scholar]




