
Translate this page into:
An Infant with Splenohepatomegaly: A Rare Cause
Address for correspondence: Dr. Vinoth N Ponnurangam, Sri Ramachandra Medical College, Porur, Chennai - 600 116, Tamil Nadu, India. E-mail: Vindoc1977@gmail.com
-
Received: ,
Accepted: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Osteopetrosis is a rare congenital disorder of bone resorption, caused by failure of osteoclasts to reabsorb immature bone. Malignant infantile osteopetrosis presents in early life with generalized osteosclerosis and decreased bone marrow spaces, resulting in anemia, splenohepatomegaly due to extramedullary hematopoiesis, cranial nerve compression, and growth failure. It is a fatal condition with death occurring within the first year of life. Bone marrow transplant remains the only curative treatment. We present a report of an infant with splenohepatomegaly, who was diagnosed with malignant infantile osteopetrosis.
Keywords
Bone marrow transplantation
osteopetrosis
osteosclerosis
splenohepatomegaly

INTRODUCTION

Osteopetrosis is an inherited disorder of the bones. Bones appear radioopaque and are characterized by increased cortical thickness and reduced marrow space. The clinical syndrome of osteopetrosis is characterized by abnormal bone modeling and remodeling due to failure of osteoclastic bone reabsorption. This results in skeletal fragility in spite of increased bone mass, hematopoietic insufficiency, which is a result of decreased marrow space, nerve entrapment syndromes, growth retardation, and disrupted tooth eruption. We report an infant with splenohepatomegaly, who was diagnosed with malignant infantile osteopetrosis.
CASE REPORT
A 45-day-old female baby, second born to non-consanguineous parents, presented with abdominal distension and skin rash of 2 weeks duration. The baby had no significant history related to this illness. Her older sibling died of unknown etiology at 3 months of age. Anthropometric measurements of the baby were normal for her age. On examination, there was pallor and petechiae, hepatomegaly (edge 4 cm below the right costal margin), and splenomegaly (edge 7 cm below the left costal margin). No other body systems appeared to be involved. Ophthalmic examination including fundus was normal. Her hemoglobin was 6.8 gm% (normal range: 11-14 gm%), total white blood cell count was 43,900 cells/mm3 (normal range: 5000-17,000 cells/cu.mm3) with a differential count of polymorphonuclear cells 30%, lymphocytes 57%, eosinophils 3%, and monocytes 9%, and platelets numbered 30,000/mm3 (normal range: 2-4.9 × 100,000/mm3). Peripheral smear showed leukoerythroblastosis. Reticulocytes were 6% (0.1-2.4%). Liver and renal function tests were within normal limits. Her serum calcium was 9.0 mg/dl (normal range: 8.5-10.1 mg/dl), phosphorus 3.7 mg/dl (normal range: 2.6-4.9 mg/dl), alkaline phosphatase 370 U/l (normal range: 54-369 U/l), and lactate dehydrogenase (LDH) was 640 U/l (normal range: 180-435 U/l). Retroviral studies and TORCH titers were negative and cultures were sterile. Abdominal ultrasonogram confirmed splenohepatomegaly. On Day 3 of admission, the child developed cough, hence X-ray chest was taken. The X-ray of the skeleton showed bone in bone appearance in femur, tibia, humerus, radius, ulna, and iliac bones [Figure 1]. Baby was diagnosed as having an autosomal recessive form of osteopetrosis [malignant infantile osteopetrosis (MIOP)]. She was treated with 3% normal saline nebulizations for bronchiolitis, blood transfusion, calcium and vitamin D supplements. Parents were counseled regarding the diagnosis, the need for bone marrow transplant and advised to have regular follow-up in the outpatient department. However, the child was lost to follow-up.
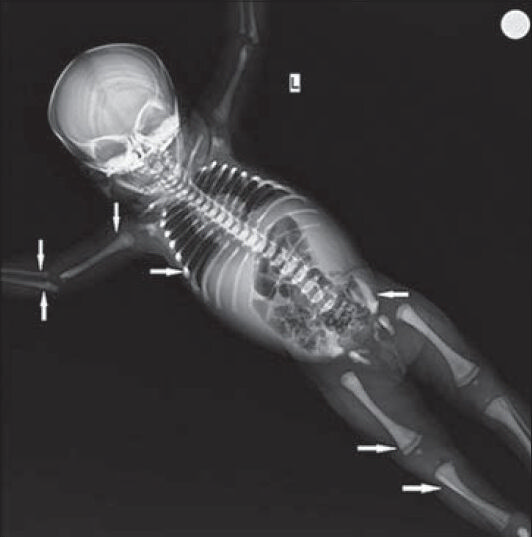
- 45-dayold infant presented with splenohepatomegaly, which was subsequently diagnosed as malignant infantile osteopetrosis. X-ray of the skeleton shows (solid arrow) bone in bone appearance in the femur, tibia, humerus, radius, ulna, and iliac bones.
DISCUSSION
Osteopetrosis is a rare congenital genetic disorder characterized by increased bone density due to impaired osteoclastic bone reabsorption.[1] Two main forms of osteopetrosis have been described: A mild, autosomal dominant form, which is usually diagnosed incidentally or presents with mild symptoms in late childhood or adult life with good long-term survival,[2] and a severe autosomal recessive form also known as MIOP. MIOP is a fatal disorder with an incidence of 1:200,000-1:300,000.[1] In most of the patients, the recessive form occurs due to mutations in the gene encoding an osteoclastic specific subunit of the vacuolar proton pump, thereby causing disturbances in acidification needed for normal osteoclast function.[3] Most patients present with symptoms within the first year of life. Studies indicate varying age of presentation, ranging from 15 days to 18 months.[4] Diagnosis is mainly based on clinical, hematological factors and classic radiological images showing increased bone density. Initial radiographs reveal diffuse bone sclerosis and later films show the characteristic bone within a bone appearance. Computed tomography scan can be used for diagnosis, to determine the treatment outcome, and to assess the optic and auditory canal.[5] Most of the clinical manifestations are due to failure of remodeling of growing bones. This leads to narrowing of cranial nerve foramina and encroachment on marrow spaces, which result in secondary complications, such as optic and auditory nerve dysfunction and anemia. Anemia is accompanied by compensatory extramedullary hematopoiesis in the liver and spleen leading to splenohepatomegaly.[126] Hemolysis resulting from hypersplenism worsens the anemia and thrombocytopenia. Hematological impairment occurs within the first year of life in about 75% of patients and its presence within 3 months of age is indicative of poor prognosis.[6] Typically, the infant shows psychomotor delay, failure to thrive, dental problems, pathological fractures, osteomyelitis of mandible, worsening of anemia, and cranial neuropathies. Dental symptoms include delayed tooth eruption, enamel hypoplasia, disturbed dentinogenesis, hypomineralization of enamel and dentin, propensity for tooth decay, defects of the periodontal membrane, thickened lamina dura, mandibular protrusion, and the presence of odontomas.[7] Seldom do these children survive beyond infancy. Children who do survive face learning disorders due to visual and hearing defects; however, in most of these children, the mental ability does not appear to be affected.[8] In any infant with anemia and splenohepatomegaly, osteopetrosis should be included in the differential diagnosis, along with hemolytic anemias, retroviral/TORCH infections, leukemia, and pkynodystosis. In the present case, liver function test and peripheral smear were not suggestive of hemolysis, so hemolytic anemia was excluded. As retroviral studies and TORCH titers were negative, it was unlikely to be due to infectious etiology. Leukemia was ruled out due to absence of blast cells in the peripheral smear. Bone marrow aspiration and biopsy was not done due to financial constraints. Pkynodystosis has similar phenotypic features to osteopetrosis and differs radiologically in that it shows partial agenesis/aplasia of terminal phalanges simulating acro-osteolysis, obtuse mandibular gonial angle often with relative prognathism, vertebral segmentation anomalies, particularly upper cervical and lower lumbar, and erosion of lateral aspect of the clavicles.[9] Parathormone, calcitriol, interferon gamma, and steroids have been used to treat osteopetrosis, with some benefits.[12] Supportive measures like maintaining dental hygiene, blood transfusions, and treatment of infections are of outmost importance in patients who survive beyond infancy. Bone marrow transplant is the main treatment and early transplantation reduces the risk of irreversible neurological damage that occurs during infancy. Good results have been obtained in patients who get bone marrow transplant from human leukocyte antigen (HLA)-matched sibling donors. The 5-year survival for recipients of HLA-identical bone marrow transplants is 79%.[10] A precise diagnosis is essential for early access to bone marrow transplant and also for genetic counseling of parents, as future siblings of such patients have a 25% risk of developing osteopetrosis.[12] Most of the children die during infancy or early childhood due to lack of bone marrow transplant.[4]
CONCLUSION
Osteopetrosis should be included in the differential diagnosis of any infant presenting with anemia, splenohepatomegaly, and having radiological features of bony sclerosis. Early diagnosis is crucial so that bone marrow transplantation can be done before irreversible damage occurs and genetic counseling regarding recurrence rates can be given to parents.
ACKNOWLEDGMENTS
The authors would like to acknowledge the efforts of Dr. Krithika Prabaharan, MD and Dr. Ram Mohan, MD, Junior Resident, Department of Pediatrics, Sri Ramachandra Medical College for their assistance in this case report.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2014/4/1/48/139738
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- A single-center experience in 20 patients with infantile malignant osteopetrosis. Am J Hematol. 2009;84:473-9.
- [Google Scholar]
- Novel mutations in Indian patients with autosomal recessive infantile malignant osteopetrosis. Indian J Med Res. 2010;131:508-14.
- [Google Scholar]
- Autosomal recessive osteopetrosis: Variability of finding sat diagnosis and during the natural course. Pediatrics. 1994;93:247-53.
- [Google Scholar]
- Malignant infantile osteopetrosis: Dental effects in paediatric patients. Case reports. Eur J Paediatr Dent. 2006;7:39-44.
- [Google Scholar]
- Disorders Involving Defective Bone Resorption. In: Nelson Textbook of Pediatrics (19th ed). Philadelphia, PA, USA: Saunders, Elsevier; 2011. p. :2432-3.
- [Google Scholar]
- Successful nonmyeloablative cord blood transplantation for an infant with malignant infantile osteopetrosis. J Pediatr Hematol Oncol. 2005;27:495-8.
- [Google Scholar]




