
Translate this page into:
Symmetrical Chorioretinal Colobomata with Craniovertebral Junction Anomalies in CHARGE Syndrome - A Case Report with Review of Literature
Address for correspondence: Dr. Tanie Natung, Department of Ophthalmology, North Eastern Indira Gandhi Regional Institute of Health and Medical Sciences, Shillong, Meghalaya - 793 018, India. E-mail: natungtanie@gmail.com
-
Received: ,
Accepted: ,
This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
This article was originally published by Medknow Publications & Media Pvt Ltd and was migrated to Scientific Scholar after the change of Publisher.
Abstract
CHARGE syndrome is a common cause of congenital anomalies. Its rate of incidence is about 1:10,000. It is phenotypically heterogeneous, usually a sporadic or autosomal dominant disorder resulting from a mutation in the CHD7 (chromodomain helicase DNA-binding protein) gene. Since the time it was first described by Hall,[1] the knowledge of the clinical characteristics of CHARGE syndrome has increased over the years. Recently, basiocciput hypoplasia and basilar invagination in patients with CHARGE syndrome have been reported. We report here a case of CHARGE syndrome where there is involvement of symmetrical chorioretinal colobomata with craniovertebral junction anomalies. The patient had symmetrical chorioretinal colobomata, external and inner ear anomalies, sensorineural deafness, characteristic facial appearance, retarded growth and development, history of patent ductus arteriosus, and craniovertebral junction anomalies. Craniovertebral junction anomalies may be an under-diagnosed phenotypic expression of CHARGE syndrome. The diagnostic criteria of CHARGE syndrome may require further revision to include the addition of craniovertebral junction anomalies.
Keywords
Atresia choanae
charge syndrome
CHD7
chorioretinal colobomata
craniovertebral junction anomalies
semicircular canals

INTRODUCTION

CHARGE syndrome is a well-known cause of congenital anomalies. It is a complex disorder and is phenotypically heterogeneous with multisystemic congenital anomalies. It has a reported incidence of approximately 1 in 10,000. It occurs more often due to sporadic mutations in chromodomain helicase DNA-binding protein (CHD7) but when inherited, the mutation is transferred in an autosomal dominant pattern. Originally, the acronym ‘CHARGE’ stood for coloboma, heart disease, atresia choanae, retarded growth and retarded development, and/or Central Nervous System (CNS) anomalies, genital hypoplasia, and ear anomalies/deafness. Since the time it was first described by Hall,[1] the understanding of the phenotype expression of the CHARGE syndrome has increased over the years. Recently, patients with CHARGE syndrome have been reported to show basiocciput hypoplasia and basilar invagination.[2] Here, we report a case of CHARGE syndrome where there is involvement of symmetrical chorioretinal colobomata with craniovertebral junction anomalies and present a brief review of the literature.
CASE REPORT
A 27-year-old woman presented with complaints of poor vision in both eyes and inability to hear since birth. She had undergone surgical ligation of patent ductus arteriosus (PDA) in childhood. There was no feeding difficulty in childhood. She had retarded growth and development and a characteristic facial appearance. There was bilateral Grade I/II microtia [Figure 1].
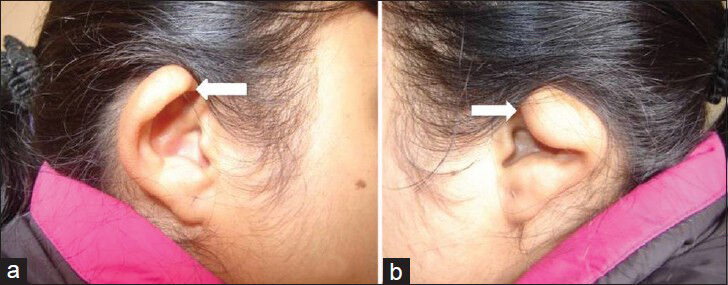
- 27-year-old female with poor vision and inability to hear was subsequently diagnosed with CHARGE syndrome. (a and b) Standard photographs of the ears from lateral view show bilateral microtia grade I/II (arrows).
Her vision was 20/2000 in both eyes. Ocular adnexa, ocular motility, and anterior segment were normal in both eyes. There was no iris coloboma but symmetrical, large colobomata involving the optic nerve, choroid, and retina were present [Figure 2a and b]. The colobomata included the macula. There were no neurological dysfunction or limb abnormalities.

- 27-year-old female with poor vision and inability to hear was subsequently diagnosed with CHARGE syndrome. (a and b) Fundus photographs (antero-posterior view) show bilateral, symmetrical, chorioretinal colobomata involving the optic discs and macula (arrows).
There was severe to profound sensorineural deafness on pure tone audiometry examination and brain stem evoked response audiometry with identifiable peak V on 125 dB stimulus in both ears. Echocardiography revealed normal cardiac chamber dimensions and valve morphology with no residual shunt across PDA closure.
Ultrasound examination of the abdomen revealed an infantile uterus. X-ray KUB (kidney, urether, and bladder) region and serum calcium were within normal limits.
Magnetic resonance imaging (MRI) revealed bilateral hypoplasia of vestibule, lateral semicircular canals, and cochlea with visualization of about 1½-turns [Figure 3a and b]. Vestibulo-cochlear nerves on both sides were of smaller size than the adjacent facial nerves with narrow internal acoustic meatus. There was basilar invagination with short clivus, fused C2 and C3 vertebrae, and partially occipitalized atlas [Figure 3c]. There was a large posterior global defect with adjacent deformed and thinned out sclera around the optic disc in both eyes [Figure 3d]. The brain was normal.
Chromosomal analysis revealed normal female karyotype (46 XX chromosomes) in all the metaphases analyzed. CHD7 gene analysis could not be done.
A diagnosis of CHARGE syndrome was arrived at by satisfying three major criteria prelisted by Blake et al., namely, i) Coloboma (retina and choroid), ii) Characteristic ear abnormalities, and iii) Cranial nerve dysfunction (sensorineural deafness), plus three minor criteria, namely, i) Cardiovascular malformations, ii) Growth deficiency, and iii) Distinctive face. The criteria for typical CHARGE syndrome according to Verloes are also met, namely, two major criteria of i) Coloboma (choroid) and ii) Hypoplastic semi-circular canals, plus three minor criteria of i) Rhombencephalic dysfunction (sensorineural deafness), ii) Abnormal external ear, and iii) Malformation of mediastinal organs (heart).
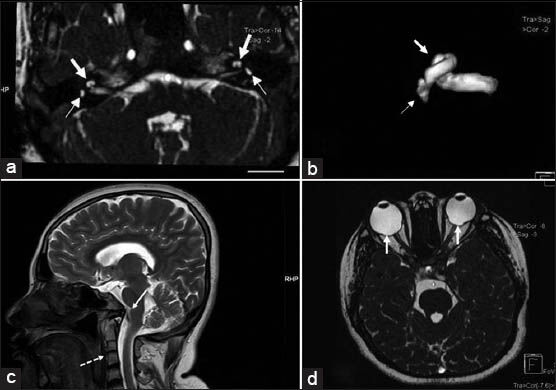
- 27-year-old female with poor vision and inability to hear was subsequently diagnosed with CHARGE syndrome. (a) Axial Constructive Interference in Steady State (CISS) T2-weighted MRI at the level of internal auditory meatus shows bilateral hypoplasia of semicircular canals seen as small saccular structures instead of normal “D” like canalicular structure (small arrows) and cochlea shows only one and half (11/2) turns instead of normal two and half (21/2) or more (large arrows). (b) Three-dimensional reconstruction of CISS MRI images of the right internal auditory apparatus confirming rudimentary semicircular canals (small arrow) and hypoplasia of cochlea (large arrow). (c) T2 Fast spin echo mid-sagittal reconstruction of MRI head shows associated craniovertebral junction anomaly with fused C2 and C3 vertebrae with rudimentary intervertebral disc (dotted arrow) and basilar invagination (solid arrow). (d) Axial CISS T2 weighted MRI at the level of orbits shows defects of choroid (solid arrows).
Prophylactic laser retinopexy was done in both the eyes along the border of the coloboma to reduce the risk of future retinal detachments. Her corrected visual acuity was 20/2000 in both eyes.
Her two sisters and mother were found to be normal.
DISCUSSION
The features related to CHARGE syndrome was first described by Hall in 17 children with choanal atresia associated with multiple congenital anomalies [MCA][1] and independently by Hittner in 10 MCA patients with coloboma, heart disease, and hearing loss.[3] Therefore, this disorder came to be known as ‘Hall–Hittner syndrome’ or ‘CHARGE association’. But it was Pagon et al., who coined the acronym ‘CHARGE’ to include the six cardinal features of coloboma, heart disease, atresia choanae, retarded growth and retarded development and/or CNS anomalies, genital hypoplasia, and ear anomalies/deafness.[4] There has been a histopathological report of symmetrical involvement of ocular colobomata in a case report of CHARGE syndrome.[5]
The phenotype expression of CHARGE syndrome has been updated over the years to include additional features and the criteria for diagnosis were further refined. Blake et al., proposed a renewed definition of CHARGE based on 11 criteria.[6] The new definition included brainstem dysfunctions as a major component. It had four major or three major + three minor diagnostic criteria. Verloes updated the definition of CHARGE further and proposed three new major and five minor signs. The major (“the 3 C”) signs included coloboma, atresia of choanae, and hypoplastic semi-circular canals. He further observed that the semicircular canal anomalies have high specificity for CHARGE. He gave the definition for typical, partial, and atypical CHARGE syndrome.[7] The evolving diagnostic criteria of CHARGE syndrome are summarized in Table 1.[4678]

The CHD7 gene has been established as a genetic etiology for CHARGE syndrome.[289] About 60-70% of patients with CHARGE syndrome have CHD7 mutations.[29] CHD7 gene analysis could not be done in our case.
Chorioretinal coloboma is present in 53-90% cases of CHARGE syndrome.[134568] Bilateral involvement is present in 70-80%.[8] Colobomata are usually bilateral and somewhat asymmetric.[10] Other ocular anomalies are iris or eyelid coloboma, microphthalmia, microcornea, strabismus, nystagmus, optic nerve hypoplasia, persistent hyperplastic primary vitreous, anophthalmos, and anisometropia.[13456810] Our case had large and symmetrical colobomata involving the optic disc, choroid, and retina. The vision was very poor due to the involvement of optic disc and macula.
Unilateral or bilateral atresia of the choanae is an infrequently occurring developmental anomaly caused by membranous and/or bony obstruction of the posterior nasal choanae.[10] Chestler et al., did not find a single case of atresia choanae in their case series.[10] Our case did not have atresia choanae or cleft lip or palate. Atresia choanae is usually present in 35-65% of cases.[468] Deafness affects 60-90% of cases.[468] The most common hearing defects are severe conductive or mixed hearing loss.[8] Our case had bilateral and asymmetrical grade I/II microtia and hypoplasia of semicircular canal and cochlea with severe to profound sensorineural hearing defect.
Congenital heart defects like atrial septal defect, endocardial cushion defect, PDA, and Tetralogy of Fallot are usually seen in CHARGE syndrome.[13468910] These defects are found in 50-85% of patients.[13468] Our patient had PDA, which was surgically ligated in her childhood.
Basioccipital hypoplasia and basilar invagination have been reported in cases of CHARGE syndrome by Fujita et al., in their case series. They identified basioccipital hypoplasia in seven of the eight patients with CHARGE syndrome. Of those, five had associated basilar invagination. The authors concluded that basioccipital hypoplasia and basilar invagination are prevalent in patients with CHARGE syndrome, although they mentioned that their case series was biased. They had suggested these anomalies as possible new diagnostic criteria.[2] Our case had basilar invagination with short clivus, fused cervical vertebrae, and occipitalized atlas. From the case series of Fujita et al., and our case, it appears that craniovertebral junction anomalies may be an underdiagnosed association of CHARGE syndrome. Therefore, if MRI of basiocciput is done routinely in cases of CHARGE syndrome, craniovertebral junction anomalies may be picked up more often. The diagnostic criteria for CHARGE syndrome may require further revision to include craniovertebral anomalies.
CONCLUSION
We have reported here a case of CHARGE syndrome where there is involvement of symmetrical chorioretinal colobomata with craniovertebral junction anomalies. Craniovertebral junction anomalies may be an underdiagnosed association of CHARGE syndrome. Its diagnostic criteria may require further revision with the addition of craniovertebral junction anomalies as one of the criteria. We suggest MRI of basiocciput along with internal auditory apparatus to be done routinely in cases of CHARGE syndrome to rule out craniovertebral anomalies.
Available FREE in open access from: http://www.clinicalimagingscience.org/text.asp?2014/4/1/5/126046
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- Abnormal Basiocciput Development in CHARGE Syndrome. AJNR Am J Neuroradiol. 2009;30:629-34.
- [Google Scholar]
- Colobomatous microphthalmia, heart disease, hearing loss and mental retardation - A syndrome. J Pediatr Ophthalmol Strabismus. 1979;16:122-8.
- [Google Scholar]
- Coloboma, congenital heart disease and choanal atresia with multiple anomalies: CHARGE association. J Pediatr. 1981;99:223-7.
- [Google Scholar]
- CHARGE Association: Histopathological Report of Two Cases and a Review. J Pediar Ophthalmol Strabismus. 1998;35:100-6.
- [Google Scholar]
- CHARGE Association: Update and Review for the Primary Pediatrician. Clin Pediatr (Phila). 1998;37:159-73.
- [Google Scholar]
- Updated diagnostic Criteria for CHARGE Syndrome: A Proposal. Am J Med Genet A. 2005;133A:306-8.
- [Google Scholar]
- Molecular and phenotypic aspects of CHD7 mutation in CHARGE syndrome. Am J Med Genet A. 2010;152A:674-86.
- [Google Scholar]
- Ocular Findings in CHARGE Syndrome. Six case reports and a review. Ophthalmology. 1988;95:1613-9.
- [Google Scholar]




